
Abstract
Mangroves are complex land-sea transition ecosystems whose microbiota are essential for their nutrient recycling and conservation. Brazil is the third-largest estuarine area in the world and “Baía de Todos os Santos” (BTS) is one of the largest bays of the country, with wide anthropogenic exploration. Using a metagenomic approach, we investigated composition and functional adaptability as signatures of the microbiome of pristine and anthropized areas of BTS, including those under petroleum refinery influence. The taxonomic analysis showed dominance of sulfate-reducing Desulfobacteraceae, Rhodobacteraceae, and Flavobacteriaceae. Taxa were significantly diverse between pristine and disturbed areas. Disturbed mangroves showed a notary increase in abundance of halophilic, sulfur-related, and hydrocarbon-degrading genera and a decrease in diatoms compared to pristine area. The metabolic profile of BTS mangroves was correlated with the differentially abundant microbiota. Two ecological scenarios were observed: one marked by functions of central metabolism associated with biomass degradation and another by mechanisms of microbial adaptability to pollution conditions and environmental degradation. Part of the microbiome was distinct and not abundant in Brazilian estuarine soils. The microbiome signature observed in each BTS mangrove reflects how human actions impact the diversity of these ecosystems and also emphasize their role in attempting to restore disturbed mangroves. The microbiome may act as a potential biological indicator of the preservation status of these soils, despite the limitation of soil property conditions. Additionally, our data pointed to metagenomics as an additional tool for environmental assessment and reinforced the need for protective measures for the mangroves under study.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mangroves are intertidal ecosystems of worldwide distribution in tropical and subtropical coastlines (Giri et al. 2011). These ecosystems have high primary productivity and are important for climate change mitigation due to their large capacity for biogeochemical nutrient cycling and increased carbon storage capacity (Alongi 2002, 2014; Bouillon et al. 2008), besides providing environmental conditions suitable to the development of marine species and contributing to area preservation against erosion and natural disasters (Palit et al. 2022). Also, they play a major role in energy and mineral cycles along tropical coasts, acting as a biogeochemical barrier to trace metal transport and a nutrient source for microorganisms, plants, and animals (Freitas et al. 2002). Regardless of their ecological importance, 35% of the global mangrove areas have been lost over the last 20 years (Valiela et al. 2001) as a consequence of strong anthropogenic pressures (Duke et al. 2007; Spalding et al. 2010; Hamilton and Casey 2016), impacting the benthic biodiversity and leading to a drastic decline of microbially mediated decomposition rates (Carugati et al. 2018).
Mangroves cover 9900 km2 of Brazilian territory, the country with the third-largest estuarine area (Giri et al. 2011), comprising 8.5% of the global mangrove area (Diniz et al. 2019). The “Baía de Todos os Santos” (BTS) is the largest bay in Brazil and the second largest in the world (IBGE 2019). Located in Northeastern Brazil, around the 8th major metropolitan area of the country (Salvador city, Bahia State), BTS has a maximum size of 1223 km2 and an average depth of 9.8 m. The intertidal area corresponds to 27% of the maximum area of the BTS (327 km2), most of which is occupied by mangroves and non-vegetable regions of varied sediment textures (Cirano and Lessa 2007). BTS holds the largest petrochemical settlement in the southern hemisphere and its maritime activity represents almost 5% of the annual flow throughout Brazil. The offshore oil and gas fields located 100 km from the bay (ANP 2017) impact the continental shelf, located northeast of the bay, through the receipt of biochemical drainage and sewage (Cirano and Lessa 2007). In addition to oil exploration, areas of the Baía de Todos os Santos have been used for the extraction of calcareous sands, fishing activity, and intensive tourism (Leao and Dominguez 2000). Moreover, a study of chemical monitoring of saline water and sediments at Aratu harbor over 2 years (2010–2012) indicated the bioaccumulation of environmental contaminants copper and lead metals in sediments of BTS (Rocha et al. 2016a, b).
Studies of microbial communities of mangroves have been highlighted due to their close nutrient-plant relationship, which functions as a mechanism to recycle and conserve nutrients essential for the productivity, conservation, and rehabilitation of these ecosystems (Holguin et al. 2001; Jansson and Hofmockel 2018). In this context, metagenomics and 16S rRNA high-throughput sequencing methodologies have been employed to evaluate the taxonomic and functional diversity of mangroves (Allard et al. 2020; Nóbrega et al. 2022), as well as community changes resulting from environmental, anthropogenic, and industry actions around the world (Andreote et al. 2012; Cabral et al. 2016; Huergo et al. 2018; Li et al. 2019; Liao et al. 2020; Mai et al. 2021).
The microbiome and their potential metabolic properties in Brazilian mangroves have been poorly characterized (40 public references on The National Center for Biotechnology Information, January 2024) and widely focused on North, Southeast, and Northeast regions, mainly in Amazonas, São Paulo, and Ceará States, respectively (Andreote et al. 2012; Nogueira et al. 2015; Mendes and Tsai 2018; Cotta et al. 2019; Nóbrega et al. 2022; Costa et al. 2023). In Amazonian mangroves, microbial groups driving carbon, nitrogen, methane, and mainly sulfur cycles and that are consistently found across pristine areas (Nóbrega et al. 2022) suffer a considerable reduction of diversity in areas under human impact (Costa et al. 2023). In Southeastern Brazil, the microbial communities of degraded mangroves are structured orderly, adapting to new conditions and establishing a functional balance without loss or impairment of the ecological system (Andreote et al. 2012; Cotta et al. 2019). In addition, these soils have an elevated concentration of heavy metals, originating from pollution by petroleum, sludge, and other urban waste, with members of Gammaproteobacteria and Deltaproteobacteria classes contributing to the detoxification processes (Cabral et al. 2016). Sanders et al. (2014) observed a rapid accumulation of organic carbon and total nitrogen in these ecosystems due to increased organic matter originating from phytoplankton, benthic algae, or other autochthonous and allochthonous sources, which can lead to coastal eutrophication. Similarly to Southeastern, in Paranaguá Bay (Southern region, Paraná State, Brazil), metabolic functions are more conserved than microbial structure, clustered according to biome type over the salinity gradient (Ceccon et al. 2019).
In the Brazilian Northeastern, the microbiome of anthropized mangroves from Ceará State varied in response to a semiarid climate with altered Proteobacterial class abundance, possibly related to eutrophication. However, they did not metabolically differ from the pristine ecosystem (Nogueira et al. 2015). Minimum temperature, precipitation, organic carbon, and evapotranspiration were the main variations in the diversity of the northeastern semiarid microbiomes. Furthermore, the resilience of these communities to cope with adverse conditions may be the factor triggering functional mangrove adaptability (Tavares et al. 2021).
In the Bahia State Brazil, the bioavailability of organic and toxic compounds affecting the geochemical cycles in mangroves has been explored (Paixão et al. 2011; Silva et al. 2014; Bomfim et al. 2015; Paes et al. 2022), but very little attention has been directed to the microbial communities inhabiting the BTS.
This metagenomic study aims to identify microbial and metabolic signatures (i.e., singular microbial community structure and functional diversity) of conserved, anthropized, and oil refinery estuaries along the BTS under influence of human and industrial activities as a possible assessment measure tool of the preservation degree from these areas.
Material and methods
Study sites and sample collection
The study covered eight distinct mangrove regions of the Baía de Todos os Santos (BTS), an inlet of the Brazilian coast in Bahia (Fig. 1 and Supplementary Table 1). A total of 96 mangrove sediment samples were collected from regions inside and outside the BTS, including the surroundings and inside the refinery. The samples were obtained in triplicate for each one of the 32 points, being Caípe (04), Mataripe (16), ETDI (03), Parque Niterói (01), Caboto (02), Passé (02), Jeribatuba (02), and Cairu (02) (Supplementary Table 1). The Caípe (CAI) and Mataripe (MAT) regions correspond to the rivers Caípe and Mataripe, respectively, which flow into the BTS and cross an oil refinery. Caípe River borders the refinery, while the Mataripe River runs through the middle of the refinery, which justifies the largest number of sampling points in this region. The ETDI (ETDI) and Parque Niterói (Pq Niterói—NIT) regions correspond to two areas inside the refinery. ETDI is an effluent treatment station where the current mangrove has been restored. However, it still has very different characteristics from a typical mangrove. In this area, the sediment was considerably finer and more profound than other BTS sampling sites, making access impossible without the aid of supports, which was not necessary at any other sampling points. Organoleptic characteristics were also markedly different from those found in other mangroves, even those under the direct influence of the refinery. Parque Niterói (NIT) is an area where the mangrove restoration has not been successful. Caboto (CBT) and Passé (PAS) areas are near the refinery and although not under its direct influence are impacted by other types of anthropogenic pressures such as other industries and human occupation. Jeribatuba (JER) and Cairu (CAM) are located in preserved areas away from great anthropic pressures and are considered reference regions within and outside the BTS, respectively.

Geographic location of Baía de Todos os Santos (BTS) mangroves sampled in this study, located around Salvador City, Bahia State, Brazil. Eight mangrove regions were selected, and the number of points analyzed in each region is shown in parentheses. Points of Cairu were named as CAM 01 up to 02; Jeribatuba as JER 01 up to 02; Parque Niterói as NIT 01; Caípe as CAI 01 up to 04; Mataripe as MAT 01 up to 16; ETDI as ETD 01 up to 03; Passé as PAS 01 up to 02; and Caboto as CBT 01 up to 02. For each point, three samples were collected
Sediments were collected on the first 10 days of October 2019 at low tide at 0 to 2 cm depth. Soil samples were collected with a sterilized stainless steel spatula, with the collector wearing a face mask and nitrile gloves. Soils were placed in RNase-free Falcon tubes and kept on ice (4 °C) until arrival at the field base. There, they were frozen at − 20 °C and later transported on dry ice to the laboratory, where they were stored in an ultrafreezer (− 80 °C) until processing.
DNA extraction and whole genome shotgun sequencing
Total DNA extraction was performed using the PowerSoil® DNA Isolation Kit (Mobio Labs, Inc., Solana Beach, CA, USA) according to the manufacturer’s guideline at SENAI Institute of Innovation in Biosynthetic and Fibers (SENAI CETIQT, Rio de Janeiro, RJ, Brazil). The quantitative and qualitative DNA analysis and metagenomic library preparation and sequencing were performed at Computational Genomics Unity Darcy Fontoura de Almeida (UGCDFA) of the National Laboratory of Scientific Computation (LNCC) (Petrópolis, RJ, Brazil). The DNA concentration and integrity were determined using Agilent TapeStation 4200 System (Agilent Technologies, USA) with Genomic DNA ScreenTape according to the manufacturer’s instructions. Libraries were constructed using the Nextera DNA Flex library preparation kit (Illumina, USA) according to the manufacturer’s recommendations. Sequencing was conducted on an Illumina NextSeq 500 platform (Illumina, San Diego, CA, USA) using the NextSeq 500/550 high-output kit v2.5 (Illumina, USA), with the system set to produce 2 × 150-bp reads.
Bioinformatic processing and statistical analyses
Raw reads were submitted to BBduk (BBMap software v.38.81 [https://github.com/BioInfoTools/BBMap]) for quality control (i.e., the identification and filtering of low-quality reads and sequencing artifacts). Reads with a quality threshold lower than a Phred score of 20 (with a sliding window of 10 bases) and a length smaller than 50 bp, Illumina adapters, and phiX174 were removed using the following parameters: minlength = 50, mink = 8, qout = auto, hdist = 1, k = 31, trimq = 10, qtrim = rl, ktrim = l, minavgquality = 20, and statscolumns = 5.
The high-quality reads were then submitted to taxonomic classification using the Kaiju 1.7.3 software (Menzel et al. 2016) and the NR_EUK database (February 2021 version). It is known that Kaiju specificity and accuracy are dependent on the coverage and taxa abundance, with limitations for more precise species-level inferences (Ye et al. 2019). Nevertheless, in simulations where Critical Assessment of Metagenome Interpretation (CAMI) datasets (i.e., short DNA sequencing reads, comprised of different taxon profiles, with only 30–40% of reads are simulated from known taxa—Sczyrba et al. 2017) were used at the genus level, Kaiju had an improvement on precision-recall curve (AUPR) scores similarly to those observed to the other classifiers (Ye et al. 2019). Thus, Kaiju was used to capture partial genomes, in a broader overview of the environmental diversity of sampling mangroves.
The Chao1 estimator investigated microbial richness, and Shannon and Simpson indexes were applied to compute the diversity under the absolute abundance of bacterial species. Richness and diversity indices were obtained using the skbio.diversity.alpha_diversity function of a Python script written in the skbio package (scikit-bio Development Team. 2020. scikit-bio: a bioinformatics library for data scientists, students, and developers, version 0.5.5). Alpha diversity indices were sampled graphically using the vegan package (version 2.6–4, R Core Team 2021). The diversity of each point was compared pairwise using ANOVA, with Tukey’s post hoc test (ggpplot2 package, version 3.4.1, R Core Team 2021), considering a value of p < 0.05 for statistical significance. Principal coordinate analysis (PcoA), generated to determine the distances or dissimilarities between the structures of the microbial communities, was obtained using the NMDS method and Bray–Curtis metric available in the Phyloseq package (McMurdie and Holmes 2013)(version 1.40.0, R Core Team 2021).
Nonrandom library size normalization corrected sequencing depth variations among samples to make the samples comparable. For this, a factor reflecting each sample-specific library size was applied to the respective read counts [calculated as factor = (n trim reads ss/n trim reads sls) × OTU reads ss, where ss is the specific sample, sls is the smallest library sample size across all samples, and OTU is the operational taxonomic units]. The microbial composition was investigated by first analyzing representative taxa (for phylum, family, genus, and species levels) and, in more detail, those that were differentially abundant (for family and genus levels). The first one was restricted to the 15 taxa with the highest abundance for each sampling point. The entire set of taxa identified for the taxonomic level was evaluated to analyze differential abundance. Sequences of prokaryotes, eukaryotes, archaea, and viruses were maintained in the analyses to broadly characterize the diversity found in the sampled mangroves, as well as identify taxa other than bacterial that could contribute as members differentially present in a specific area. The difference in microbial composition abundance among the sampling points was obtained using the ANCOM-BC package (Lin and Peddada 2020) (version 1.6.4, R Core Team 2021). Taxa with qualitative differential abundance were selected using FDR for p-value false-positive adjustment and significance of p < 0.05. The Jvenn tool (Bardou et al. 2014) was used to identify a region’s common and/or unique taxa.
For the metabolic inference analyses, the reads were assembled into contigs using Metaspades software (version 3.14) (Nurk et al. 2017), and parameter “-k 21,33,55,77.” Only contigs larger than 500 bp were included in the subsequent analyses. The contigs were then submitted to Prodigal software (v. 2.6.3) (Hyatt et al. 2010) for prediction of gene coding sequences (or “Open reading frames,” ORFs), applying the -g 1 -p meta options. Annotation and alignments were conducted with ORFs longer than 50 amino acids against the EggNOG database (Huerta-Cepas et al. 2019) (EggNOG mapper version 2.0.8 Feb 2021), which also compiles additional information from Cluster of Orthologous Genes (COG) (Tatusov et al. 2000). The following parameters were applied for the alignment: e-value ≤ 1e-5, identity > 60%, and query/subject coverage > 60%. The functional analyses were performed to identify metabolic categories associated with the spatial distribution of sampling points and correlate them with their taxonomic composition. To investigate whether the variance of the functional matrices was correlated to the spatial distribution of the samples, a permutational multivariate analysis of variance was conducted using distance matrices and the “adonis2” function from the Vegan package (Oksanen et al. 2022)(version 2.6–4, R Core Team 2021). The dispersion among the triplicates of each sample point was verified by a multivariate analysis of homogeneity of group dispersion using the “betadisper” function from the Vegan package (version 2.6–4, R Core Team 2021). Since high heterogeneity was observed among the triplicates of each point, the matrices were clustered using the “K-means clustering” method to minimize the internal variation compared to the variability between clusters. The K-means clustering was based on EggNOG (clusterEgg) matrices for the region and sampled point. To identify functions that best distinguished sample clusters from the EggNOG matrices, supervised classification models generated by recursive partitioning and regression trees were used (Dinsdale et al. 2013). Initially, a series of test/validation partitions were created using the createDataPartition function from the Caret package (version 6.0–93, R Core Team 2021). Then, the train function was used to evaluate the parameters and performance of different classification and regression methods (lda—linear discriminant analysis; rpart—recursive partitioning and regression training; knn—K nearest neighbors, and rf—random forest). Metabolic functions of the highest importance for each sample cluster were identified using the random forest method (Breiman 2001), according to accuracy and Gini metrics (Menze et al. 2009). The specificity and predictive power metrics were evaluated using the predict and confusion matrix functions. Regression trees were generated using the “rpart” function (rpart package, version 4.1–19, R Core Team 2021), obtaining a final model with minimal redundancy risk (Breiman et al. 1984).
The variables highlighted by the classification methods were additionally analyzed in multivariate ordination models (Dinsdale et al. 2013). Redundancy analysis (RDA), by function “rda” (Vegan package, version 2.6–4, R Core Team 2021), was adopted to investigate which functional categories were correlated to the spatial distribution of samples and taxonomic composition. The “ordistep” function (Blanchet et al. 2008) (Vegan package, version 2.6–4, R Core Team 2021) was adopted to determine the metabolic functions and taxa significantly related to the matrices distribution using permutational tests. All data were scaled to a single variance for the ordination analyses, thus defining a correlation analysis between the variables. The functional and taxonomic data matrices used a relative abundance of hits. The overall significance of the model was checked using ANOVA (p < 0.05) and the significance of the canonical axes was checked with the “permutest” function (Vegan package, version 2.6–4, R Core Team 2021).
The metabolic functions identified by the classification models, together with the functions with the highest contribution to the ordination model (highest scores), were progressively selected until the set of significant metabolic functions with the highest explanatory power, and lowest residual interference was reached (Borcard et al. 2011; Dinsdale et al. 2013). A generalized linear model (GLM) was used to test the statistical significance of the ordination scores of the first two components concerning the spatial distribution categories: clusterEgg, region, and sampled point. GLM model used a Gaussian distribution (F-test), verified through histogram and dispersion parameters (Borcard et al. 2011; Dinsdale et al. 2013).
Results
The microbial diversity of the mangroves
The metagenomic sequencing from eight mangroves in “Baía de Todos os Santos” (BTS) (Fig. 1 and Supplementary Table 1) yielded 1,717,124,604 raw reads. The samples CAM01MGA (from Cairu), JER02MGB (Jeribatuba), MAT04MGA (Mataripe), and NIT01MGA (Parque Niterói) generated very poor sequencing throughput and were removed from the analyses. A total of 1,594,536,856 high-quality reads were obtained from 92 samples, of which an average of 51.68% were assigned to a taxon (Supplementary Table 2).
The microbial richness and alpha diversity of the BTS mangrove sediment samples, grouped by region or sampling points, were measured by Chao1, Shannon, and Simpson indices (Fig. 2). Mangroves under refinery influence, such as NIT, ETDI, and MAT, showed the highest estimated richness compared to those described as a reference inside (JER) and outside the BTS (CAM) (Fig. 2A). Mangroves under refinery influence also showed the highest diversity index (Fig. 2A) (Supplementary Table 3). Caboto (CBT) and Passé (PAS), which suffer indirect effects of anthropogenic actions, showed higher species richness compared to JER and CAM (Fig. 2A). However, we observed a lower diversity in the anthropized mangroves (Fig. 2A). A heterogeneity of diversity was observed among sampling points, mainly for NIT, ETDI, and MAT (p < 0.05) (Supplementary Table 3). Shannon variations were significant for some comparisons between intra-regional replicates (Fig. 2A), such as for MAT and CAM (p < 0.05) (Supplementary Table 3). Interestingly, NIT showed high diversity (p < 0.05) (Supplementary Table 3), as well as high species dominance, even with only one sampling point (Fig. 2A). The beta diversity analysis revealed that NIT had a microbiota closer to that of ETDI and both have differences in species composition compared to the other samples (Fig. 2B).

A Alpha diversity comparison of the bacterial compositions among the eight mangroves from the BTS area, measured according to the Chao1, Shannon, and Simpson indices. Boxes represent the interquartile ranges (IQRs) between the first and third quartiles (25th and 75th percentiles, respectively), and the line inside denotes the median. Whiskers indicate the lowest and the highest values within a range of 1.5-fold and the IQRs from the first and third quartiles, respectively. B Beta-diversity comparison between inter-studied regions and among the sampling points from each BTS mangrove region
Microbial composition and differential abundance
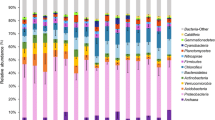
The results show an abundance of taxa belonging to the bacterial phyla Thermodesulfobacteriota (Desulfobacteria class), Pseudomonadota (from Gamma- and Alphaproteobacteria class), Bacteroidetes (mainly from Bacteroidia and Flavobacteria class), and Planctomycetes (Phycisphaerae class), followed by the Actinobacteria and Chloroflexi phyla. The Cyanobacteria phylum was identified mainly in the ETDI region (Supplementary Fig. 1). Desulfobacteraceae, Rhodobacteraceae, Flavobacteriaceae, Halieaceae, and Chromatiaceae were the most representative families in the study area (Supplementary Fig. 2).
The differential abundance (DA) analyses showed that 41 families were shared among all mangroves (p < 0.05) (Fig. 3A, B, Supplementary Table 4). An increase in abundance variation (positive log fold change—LFC) of hydrocarbon-degrading Alcanivoracaceae and purple nonsulfur Rhodospirillaceae, both Pseudomonadota phylum, was observed in mangroves inside (ETDI and NIT, LFC 0.775 and 0.669, respectively) and permeating the refinery (MAT, LFC 0.379 and 0.504, respectively) (Fig. 3A, B). An apparent decrease of more than twofold in abundance (negative log fold change) of diatoms from distinct families was observed in different mangrove sets, such as Biddulphiaceae and Odontellaceae (in mangroves inside—ETDI and NIT; and permeating the refinery—MAT); and Rhizosoleniaceae, Skeletonemataceae, and Thalassiosiraceae (in mangroves around, permeating- and those inside- the refinery) (Fig. 3A, B). Compared to CAM, which is the preserved and referenced mangrove outside the BTS, all mangroves had a decreased abundance in Hemidiscaceae and Coscinodiscaceae diatoms families, less evidenced in JER, the reference inside the BTS (Fig. 3A, B).

Differential abundance (DA) analysis of microbial composition identified in BTS mangroves for family taxonomic level. A Venn representation of common and exclusive families with significant DA in each studied area. B Representation of abundance variation of common families identified in the BTS mangroves, showing the highest log fold change (LFC) variations (positive and negative) for the area. C The highest LFC variations of families whose p-value showed DA significance only for one region. This analysis was conducted on disturbed mangroves compared to Cairu (CAM), the pristine mangrove, the reference outside the BTS. ETDI (ETD) and Parque Niterói (NIT) points were assigned as “inside refinery”; Caípe (CAI) as “around refinery”; Mataripe (MAT) points as “permeating refinery”; Caboto (CBT) and Passé (PAS) as those under “anthropogenic influence”; and Jeribatuba (JER) as “inside BTS ref.”
Among the families present in one or more samples, but which differential abundance was significant for a specific sampling group, 56 were highlighted in ETDI and NIT (Fig. 3A and C, Supplementary Table 3). At the same time, 35 were significant for Caboto and Passé, both anthropized mangroves (Fig. 3A and C, Supplementary Table 4). For instance, ETDI and NIT had a significant abundance of bacterial families Immundisolibacteraceae (LFC 3.33) and Porticoccaceae (LFC 1.81) (both Gammaproteobacteria), as well as Sulfurovaceae (Epsilonproteobacteria) (LFC 1.84); and reduction of Vaucheriaceae and Stephanodiscaceae diatoms families (LFC − 2.96 and − 2.86, respectively). CBT and PAS highlighted the occurrence of families from the Caudovirales virus order and the Puniceicoccaceae bacteria (Fig. 3A and C, Supplementary Table 4).
Analysis at lower taxonomic levels, such as genus and species, showed that the majority of the genera found in the studied mangroves belong to the Bacteria domain, being the most abundant: Gemmatimonas (Gemmatimonadetes phylum); Pseudomonas (belonging to the Pseudomonadota phylum and Gammaproteobacteria class); Streptomyces (Actinomycetota phylum); Halioglobus (Pseudomonadota phylum and Gammaproteobacteria class), and Desulfosarcina (Thermodesulfobacteriota phylum and Desulfobacteria class) (Supplementary Fig. 3). Among the species, the Gammaproteobacteria bacterium, Deltaproteobacteria bacterium, Chloroflexi bacterium, Chromatiales bacterium, and Acidobacteria bacterium were the most abundant (Supplementary Fig. 4). A high intra-variability in microbial composition was observed at the genus and species levels among the sampling points of each region studied (Supplementary Figs. 3 and 4).
In the differential abundance analysis conducted for the Bacteria domain and comparing all mangroves to the CAM mangrove (reference outside of BTS) (Fig. 4A), the refinery-impacted mangroves (ETDI and NIT) showed Immundisolibacter and Candidatus Macondimonas genera with the highest abundance ratio variations (LFC > 4) between those differentially significant. Parque Niterói differed from ETDI by the abundance of PAH-degrading Polyclyclovorans, the genus related to sulfur oxidation Thiobacillus, and Tepidicella. In addition, ETDI has a predominance of actinomycete Smaragdicoccus, the Rhodospirillaceae member Pelagibius, and the sulfur-oxidizing bacteria Sulfurovum. Moreover, Geminocystis, a genus of Cyanobacteria, was highlighted in ETD03 (Fig. 4A).

Differential abundance (DA) analysis of microbial composition identified in BTS mangroves at genus taxonomic level. A Genus with the highest log fold change values (positive and negative FLC) with p-value < 0.05 for the Bacteria domain. B Archaeal genus with the highest log fold change values (positive and negative FLC) with p-value < 0.05. No archaeal genus with significant DA was observed at Caboto (CBT) points. This analysis was conducted on mangroves inside the BTS, compared to Cairu (CAM), the pristine mangrove, and the reference outside the BTS. Points of each region were numbered, being those of ETDI designed as “ETD”; Caípe as “CAI”; Mataripe as “MAT”; Caboto as “CBT”; Jeribatuba as “JER,” and Passé as “PAS”
In the anthropized areas of PAS and CBT, an increase in the abundance of Coraliomargarita and Lentimonas (both belonging to the Puniceicoccaceae family) was evidenced. JER, a reference inside the BTS, showed a high occurrence of Aestuariivitta (Rhodobacteraceae family) and a reduction in the abundance of Petrocella (Vallitaleaceae family) and Oleiagrimonas (Rhodanobacteraceae family) (LFC < − 4) (Fig. 4A). Interestingly, Oleiagrimonas showed a positive log fold change in one point of MAT (MAT12). Mataripe, which permeates the refinery, showed a distinct profile of genera occurring in greater and lesser abundance than other areas. Roseobacter, Halochromatium, and Lamprobacter were abundant, mainly in the CAI area around the refinery. Otherwise, most sampling mangroves showed a reduction of sulfate-reducing Desulfobacter (LFC < − 2), except in PAS and JER (Fig. 4A).
For Archaea, the CAI area showed an abundance of halophilic genera Haloarchaeobius, Halogranum, and Halonotius (Fig. 4B). A diminished abundance of ammonia-oxidizing Nitrosopumilus and Candidatus Nitrosomarinus was found in the majority of mangroves. Additionally, the methanogenic archaeon Candidatus Methanoliparum was reduced in the JER area and increased in ETDI (Fig. 4B).
Metabolic structure and taxa-functional correlation in BTS mangroves
The metagenomic assembly generated around 122,830,790 contigs, with an average length of 400,803,833 bp. The N50 statistics showed that more than 50% of contigs were longer than 545,661 bp. Gene prediction resulted in 15,569,562 ORFs. The median of coding sequences varied from 6.58 to 18.97% per sample (Supplementary Table 2).
The annotation of ORFs according to the EggNOG database showed that the most abundant functional COG categories throughout the study area were energy production and conversion (C) (10.24%); amino acid transport and metabolism (E) (8.21%); cell wall/membrane/envelope biogenesis (M) (5.86%); translation, ribosomal structure, biogenesis (J) (5.58%); and transcription (K) (5.21%) (Fig. 5). Little functional variation was observed between sampling points. Of the 383 COG categories identified, 19% were common to all sampling points and triplicates and 36% occurred in at least 50% of the total 92 samples. Significant COG variations were found at the points: Caípe (CAI02, CAI04), Caboto (CBT02), ETDI (ETD02, ETD03), and Mataripe (MAT01, MAT02, MAT06, MAT07, MAT08, MAT11, MAT12, MAT13, and MAT15).

Relative abundance of the predominant metabolic categories in the points of BTS mangroves according to COG (Clusters of Orthologous Genes) from the EggNOG database
The K-means clustering partitioning method was adopted since a high heterogeneity among the triplicates variance was observed. The k = 11 showed the best clustering variance, with sample groupings more internally homogeneous and distinct from each other (Fig. 6). ETD points were the most metabolically diverse, generating two clusters, one composed of ETD03 triplicates (B and C) (cluster G) and another by ETD02 point plus ETD03 A (cluster I) (Fig. 6).

K-means clustering analysis of COG categories from the EggNOG database. According to the metabolic functions identified in the studied mangroves, the samples were grouped in 11 clusters (named A to K), each showing differences in COG categories
The distribution of functional categories significantly correlated to the sampling clusters and microbial taxa distribution (PERMANOVA p = 0.001) (Supplementary Table 5). The multivariate redundancy analysis (RDA) explained 35% of the total variance (RDA1 12%, RDA2 23%) and the highest metabolic contributions to each cluster are highlighted (Fig. 7).

Redundancy analysis (RDA) to COG categories from the EggNOG database. Arrows show the strengths and contributions of variables to each K-means cluster. Sampling points are highlighted in black, COG categories in red, and taxonomic composition at the genus level in blue. The K-means groups are shown in bold
The RDA upper left square contains mainly ETD and NIT points inside the refinery. The highest metabolic contributions to the ETD were cell motility (N) and replication, recombination, and repair (L) (for cluster G), while metagenomic sequences related to the membrane and intracellular trafficking secretion (MU) explained the greatest variations in both ETDI (ETD02 and ETD03) and NIT (cluster I). These functional groups were most strongly correlated to Immundisolibacter, Candidatus Macondimonas, and Smaragdicoccus hydrocarbon-degrading bacteria and also to sulfur-related bacteria Sulfurovum. Points of refinery-disturbed mangroves (ETDI—ETD03) around and permeating the refinery (Caípe—CAI01, CAI03, and Mataripe—MAT14 and MAT15) (clusters A, F, and D) were mainly associated with metabolic mechanisms involved in signal transduction mechanisms (T) and/or cell motility (NT, N), which are correlated to Williamwhitmania and Desulfobacter genera (Fig. 7).
At the RDA upper right square (Fig. 7), we observed two metabolic scenarios, one marked by central metabolism activity and another by biotransformation processes and bioconversion of organic and toxic components. In the first one, we highlighted the functions: amino acid transport and metabolism; carbohydrate transport and metabolism; inorganic ion transport and metabolism (EGP), mainly linked to the ETD01 point (cluster J). This correlated specifically with the point MAT01 of Mataripe (cluster K) by the contribution of metagenomic sequences related to amino acid and carbohydrate (EG) metabolism. These clusters showed a strong correlation with the occurrence of aerobic marine bacteria Pelagibius and the halophilic archaea genus Haladaptatus. Otherwise, in the second scenario, lipid transport and metabolism (I), secondary metabolites biosynthesis, transport and catabolism (Q), and defense mechanisms (V) were the main categories most correlated to the sampling points of Mataripe (MAT02, MAT03, MAT04, MAT08), Caipe (CAI02), and Jeribatuba (JER01) (clusters C and E). These variations were associated with halophilic microorganisms, such as the archaeal genera Halarchaeum and Halocalculus, and the bacterial genus Egicoccus.
The lower left square of RDA was composed of the majority of points from anthropized regions (Passé—PAS01 and PAS02; Caboto—CBT02) and the mangrove permeating the refinery (Mataripe—MAT13 and MAT16) (clusters A, B, and C). These regions were mainly associated with distinct metabolic mechanisms, such as energy production and conversion/secondary metabolites biosynthesis transport and catabolism (CQ); cytoskeleton (Z); and nucleotide transport and metabolism (F). These functional contributions were given mainly by the sulfur bacteria Thiomargarita and the moderately halophilic bacteria Pontibacterium (Fig. 7).
In some points from mangroves around and permeating the refinery (Caípe—CAI04 and Mataripe—MAT 07 and MAT09), at a lower right square of RDA, the occurrence of the bacterial genus Fodinicurvata was notary, as well as the archaeal Candidatus Nitrosomarinus and Candidatus Nitrosopelagicus, and the bacterial genus Coraliomargarita in lower contribution. These genera were associated with amino acid transport and metabolism; coenzyme transport and metabolism (EH); amino acid transport and metabolism, and intracellular trafficking, secretion, and vesicular transport (U).
Discussion
The microbial community living in tropical and subtropical mangroves has an important role in the organic matter decomposition and the recycling of nutrients of mangroves, maintaining the high ecological diversity and productivity of these ecosystems (Holguin et al. 2001). Bacteria and fungi are the major constituents of total microbial biomass in tropical mangroves (Alongi 1988). The microbial metabolic activity decreases the nutrient-deficient of these habitats through different ecological functions, such as in the carbon cycle, nitrogen fixation, anoxygenic photosynthesis, phosphate-solubilization, and sulfate-reduction (Thatoi et al. 2013).
A systematic meta-analysis of published studies on microbial diversity of different mangrove conditions pointed out that the microbial composition of these soils is mostly sulfur-related (Lai et al. 2022). Despite the sulfate-reducing Desulfobacteraceae, the microbial composition of mangroves from Baía de Todos os Santos showed a predominance of members even previously described in other Brazilian mangroves, such as Flavobacteriaceae and Rhodobacteraceae. Flavobacteriaceae was reported in anthropized soil from Ceará and Bahia States, both located in the Northeastern region (Rocha et al. 2016a, b), while Rhodobacteraceae was abundant in disturbed mangroves from São Paulo State, Southeastern region (Andreote et al. 2012). Besides, the differential abundance analysis conducted for BTS sampling areas revealed distinct microbial profiles of those previously reported in mangroves from Brazil (Nogueira et al. 2015). The disturbed mangroves from BTS exhibited higher microbial diversity than preserved sampling areas, which was also observed by De Santana et al. (2021) and Lai et al. (2022). We found a wide diversity of sulfate- and sulfur-reduction bacteria in the mangroves from BTS. Taketani et al. (2010) showed that sulfate- and sulfur-reduction bacteria microorganisms are naturally selected in pristine mangrove sediments due to their usual deficit of oxygen and the abundance of organic matter, whose alteration significantly decreases the microbial diversity. This can explain the reduction of Desulfobacter in the BTS mangroves, particularly those located inside the refinery area, compared to the pristine mangrove (reference outside of BTS). Li et al. (2019) inferred that degradation of organic contamination triggered by sulfate-reducing conditions could contribute to the enrichment of sulfate-reducing bacteria as a possible effect of bioremediation in mangroves.
In anthropized mangroves, we highlighted biomass degradation species, with the occurrence of Puniceicoccaceae, a family belonging to the Verrucomicrobia phylum, ubiquitous in the ocean, and also of viruses from the Caudovirales order. Coraliomargarita spp. (a member of Puniceicococcaceae) was abundant in BTS. Doherty et al. (2017) have suggested that these microorganisms can be specialized in the degradation of plant biomass and exudates by containing genes for sulfatases, α-L-fucosidases, and β-agarases. Soil disturbances cause an increase in organic matter oxidation and a reduction of the soil structure by degrading the loss in soil aggregates (Punia and Bharti 2023), which can explain the elevated abundance of species involved in biomass degradation in anthropized mangroves here studied. Moreover, in cases of advanced disturbance, mangrove sediments can lose up to 80% of their potential to degrade/utilize carbon, decreasing the ability of the system to perform organic matter degradation and convert primary production into biomass (Carugati et al. 2018). Additionally, an uncharacterized diversity for Caudovirales virus was reported in mangrove soils, most of them phylogenetically distant to known reference sequences and formed three clades within Sipho- and Podoviridae families (Jin et al. 2019). Viruses can significantly influence local and global biogeochemical cycles, acting in carbon decomposition in mangroves by recycling complex polysaccharide (Jin et al. 2019; Cao et al. 2022). Lelchat et al. (2019) experimentally demonstrated that marine phages can display active polysaccharides in bacterial EPS, hypothesizing that viruses could also contribute to the degradation of marine organic matter and modify its bioavailability. The microbiota of mangroves around the refinery was characterized by the occurrence of halophilic and adapted to fluctuating salinity conditions genera belonging to the Chromatiaceae family (Caumette et al. 1988) and of wide distribution in various hypersaline environments (Kim et al. 2011; Mori et al. 2016). Mangroves from BTS that permeated the refinery were associated with extremely halophilic Archaea, which correlated to lipid metabolism. The lipid metabolism of halophilic Archaea has been related to the acquisition of precursors for membrane backbones, such as those derived from the catabolization of glycerol, which are abundantly produced by green algae in response to osmotic pressure (Falb et al. 2008). In the studied mangroves permeating the refinery, we also emphasized the presence of Fodinicurvata, a bacterial genus of Rhodospirillaceae family, described as hydroxybutyrate-producer, a biodegradable polyester (Wang et al. 2009).
BTS mangroves inside and permeating the refinery, on the other hand, showed a wide and elevated occurrence of the hydrocarbon degradation microorganisms, especially of Gammaproteobacteria (such as a member of the Alcanivoraceae family and members of the genera: Immundisolibacter, Candidatus Macondimonas, Polycyclovorans, Oleiagrimonas), the Burkholderiales Tepidicella, and the Actinomycete Smaragdicoccus.
Members of the Alcanivoracaceae family, when in unpolluted areas, can consume the natural hydrocarbons, such as isoprene, produced by marine phytoplankton (Coulon et al. 2012). Immundisolibacter cernigliae, the single described species of the genus, was isolated from aerobic bioreactor-treated soil from the USA and is associated with PAH degradation (Corteselli et al. 2017). Representatives of the Candidatus Macondimonas have been described as highly abundant in contaminated coastal marine sediments and play a key ecological role in response to global oil spills (Karthikeyan et al. 2019). Additionally, the genus Polycyclovorans contains species isolated from marine diatoms and enrichment in the presence of aromatic hydrocarbons (Gutierrez et al. 2013). Oleiagrimonas, a genus reported in oil-polluted saline soil, contains species whose extracellular polymeric substances may contribute to bioremediation (Huang et al. 2015).
The predominance of hydrocarbon degradation microorganisms found in the BTS mangroves under the influence of the refinery is mainly associated with defense and secretion mechanisms. Cell motility is an important adaptation of heterotrophic bacteria and archaea to exploit resources, enhance bacteria-organic-matter coupling, and escape from unfavorable conditions (Grossart et al. 2001). Curiously, Williamwhitmania, which was associated with our data on ETDI region and the functional category cell motility, has an unusual gliding motility by external organelles (Pikuta et al. 2017). Secretion of extracellular polymeric substances (EPS) is another strategy of microbial defense. Their positive influence on the bioremediation process against the PAH contamination was demonstrated by Ali et al. (2022). The EPS secretion coincides with an increase in reactive oxygen production, which accumulation could cause damage to biomolecules and be lethal to marine species (Morris et al. 2022).
Chen et al. (2018) argue the deleterious effects of petroleum exploration, with a significant increase of PAH and total organic carbon after the oil production and construction of the Mataripe refinery (1939–1950). However, features of mangroves such as high productivity, abundant detritus, and rich organic carbon make it a preferential site for the uptake and accumulation of pollutants such as PAHs (Bernard et al. 1996). The oxidizing and mineralizing of hydrocarbons by microorganisms strongly selected in impacted environments could then act to prevent the PAH-accumulation in the sea (Duran and Cravo-Laureau 2016) and their genotoxic effects in marine sediment species (Nikolaou et al. 2009; Munoz and Albores 2011), contributing to the restoration of the mangroves.
In August 2019, the Brazilian Northeast was the target of a large-scale environmental disaster due to oil spill contamination, including the state of Bahia and the surroundings of Bahia de Todos os Santos (IBAMA 2020; Silva et al. 2021). Our data allowed us to assess the microbial community and biological processes of mangroves from Baía de Todos os Santos, Brazil, as an investigative overview of the protection status of these estuaries, especially of those disturbed areas. The anthropized estuaries showed species related to biomass degradation associated with central metabolism pathways. Differentially from pristine and anthropized areas, BTS mangroves under the influence of the refinery presented a species enrichment indicative of impacted sediment, despite the limitation of soil properties conditions. These specialized microbiota, notably distinct and not previously abundant in Brazilian estuarine soils, are adapted to the PAH increase and are highly associated with cell signaling processes. These data suggest that these species are in an active process of responding and adapting to the environment, contributing to the recovery of disturbed mangroves. Finally, our data point that metagenomics can be used as an additional and predictive environmental assessment tool and reinforce the need for constant monitoring practices to preserve the Brazilian mangroves.
Data availability
The SRA files of metagenomic raw paired-end reads for this study were deposited at the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/), under accession number PRJNA954358.
References
Ali M, Song X, Ding D, Wang Q, Zhang Z, Tang Z (2022) Bioremediation of PAHs and heavy metals co-contaminated soils: challenges and enhancement strategies. Environ Pollut 295:118686. https://doi.org/10.1016/j.envpol.2021.118686
Allard SM, Costa MT, Bulseco AN, Helfer V, Wilkins LGE, Hassenrück C, Zengler K, Zimmer M, Erazo N, Mazza Rodrigues JL, Duke N, Melo VMM, Vanwonterghem I, Junca H, Makonde HM, Jiménez DJ, Tavares TCL, Fusi M, Daffonchio D, Duarte CM, Peixoto RS, Rosado AS, Gilbert JA, Bowman J (2020) Introducing the mangrove microbiome initiative: identifying microbial research priorities and approaches to better understand, protect, and rehabilitate mangrove ecosystems. mSystems 5(5):e00658-20. https://doi.org/10.1128/mSystems.00658-20
Alongi DM (1988) Bacterial productivity and microbial biomass in tropical mangrove sediments. Microb Ecol 15:59–79. https://doi.org/10.1007/BF02012952
Alongi DM (2002) Present state and future of the world’s mangrove forests. Environ Conser 29:331–349. https://doi.org/10.1017/S0376892902000231
Alongi DM (2014) Carbon cycling and storage in mangrove forests. Ann Rev Mar Sci 6:195–219. https://doi.org/10.1146/annurev-marine-010213-135020
Andreote FD, Jiménez DJ, Chaves D, Dias ACF, Luvizotto DM, Dini-Andreote F et al (2012) The microbiome of Brazilian mangrove sediments as revealed by metagenomics. Plos One 7:e38600. https://doi.org/10.1371/journal.pone.0038600
ANP Agência Nacional do Petróleo, Gás Natural e Biocombustíveis (2017) http://www.brazil-rounds.gov.br/geral/mapas/JEQ_CAL.pdf . Accessed 22 February 2023
Bardou P, Mariette J, Escudié F, Djemiel C, Klopp C (2014) Jvenn: an interactive Venn diagram viewer. BMC Bioinforma 15:293. https://doi.org/10.1186/1471-2105-15-293
Bernard D, Pascaline H, Jeremie JJ (1996) Distribution and origin of hydrocarbons in sediments from lagoons with fringing mangrove communities. Mar Pollut Bull 32:734–739. https://doi.org/10.1016/0025-326X(96)00034-3
Blanchet FG, Legendre P, Borcard D (2008) Forward selection of explanatory variables. Ecology 89:2623–2632. https://doi.org/10.1890/07-0986.1
Bomfim MR, Gonzaga Santos JA, Vinhas Costa O, Otero JL, da Silva Vilas Boas G, da Silva-Capelão V, de Souza dos Santos E, Soledade Nacif PG (2015) Genesis, characterization, and classification of mangrove soils in the Subaé River Basin, Bahia, Brazil. Rev Bras Ciênc Solo 39:1247–1260. https://doi.org/10.1590/01000683rbcs20140555
Borcard D, Gillet F, Legendre P (2011) Numerical ecology with R. In: Numerical ecology with R. Springer, New York, NY, p 306. https://doi.org/10.1007/978-1-4419-7976-6
Bouillon S, Borges AV, Castañeda-Moya E, Diele K, Dittmar T, Duke NC et al (2008) Mangrove production and carbon sinks: a revision of global budget estimates. Glob Biogeochem Cycles 22:GB2013. https://doi.org/10.1029/2007GB003052
Breiman L (2001) Random forests. Mach Learn 45:5–32. https://doi.org/10.1023/A:1010933404324
Breiman L, Friedman JH, Olshen RA, Stone CJ (1984) Classification and regression trees. In: Classification and regression trees. Routledge, New York, p 368. https://doi.org/10.1201/9781315139470
Cabral L, Júnior GVL, Pereira de Sousa ST, Dias ACF, Lira Cadete L, Andreote FD, Hess M, de Oliveira VM (2016) Anthropogenic impact on mangrove sediments triggers differential responses in the heavy metals and antibiotic resistomes of microbial communities. Environ Pollut 216:460–469. https://doi.org/10.1016/j.envpol.2016.05.078
Cao MM, Liu SY, Bi L, Chen SJ, Wu HY, Ge Y et al (2022) Distribution characteristics of soil viruses under different precipitation gradients on the Qinghai-Tibet Plateau. Front Microbiol 13:848305. https://doi.org/10.3389/fmicb.2022.848305
Carugati L, Gatto B, Rastelli E, Martire ML, Coral C, Greco S, Danovaro R (2018) Impact of mangrove forests degradation on biodiversity and ecosystem functioning. Sci Rep 8:13298. https://doi.org/10.1038/s41598-018-31683-0
Caumette P, Baulaigue R, Matheron R (1988) Characterization of Chromatium salexigens sp. nov., a halophilic Chromatiaceae isolated from Mediterranean salinas. Syst Appl Microbiol 10:284–292. https://doi.org/10.1016/S0723-2020(88)80014-6
Ceccon DM, Faoro H, Cunha LP, de Souza EM, de Oliveira PF (2019) Metataxonomic and metagenomic analysis of mangrove microbiomes reveals community patterns driven by salinity and pH gradients in Paranaguá Bay, Brazil. Sci Total Environ 694:133609. https://doi.org/10.1016/j.scitotenv.2019.133609
Chen A, Northcross AL, Folger S (2018) Environmental degradation in Baía de Todos os Santos, Brazil: a review of the evidence. Poster Abstract. GW Annual Research Days, United States. https://hsrc.himmelfarb.gwu.edu/cgi/viewcontent.cgi?article=1608&context=gw_research_days
Cirano M, Lessa GC (2007) Oceanographic characteristics of Baía de Todos os Santos. Brasil. Rev Bras Geofís 25:363–387. https://doi.org/10.1590/S0102-261X2007000400002
Corteselli EM, Aitken MD, Singleton DR (2017) Description of Immundisolibacter cernigliae gen. nov., sp. nov., a high-molecular-weight polycyclic aromatic hydrocarbon-degrading bacterium within the class Gammaproteobacteria, and proposal of Immundisolibacterales ord. nov. and Immundisolibacteraceae fam. nov. Int J Syst Evol Microbiol 67:925–931. https://doi.org/10.1099/ijsem.0.001714
Costa GMD, Costa SS, Baraúna RA, Castilho BP, Pinheiro IC, Silva A, Schaan AP, Ribeiro-Dos-Santos GDAD (2023) Effects of degradation on microbial communities of an Amazonian mangrove. Microorganisms 11(6):1389. https://doi.org/10.3390/microorganisms11061389
Cotta SR, Cadete LL, van Elsas JD, Andreote FD, Dias ACF (2019) Exploring bacterial functionality in mangrove sediments and its capability to overcome anthropogenic activity. Mar Pollut Bull 141:586–594. https://doi.org/10.1016/j.marpolbul.2019.03.001
Coulon F, Chronopoulou PM, Fahy A, Païssé S, Goñi-Urriza M, Peperzak L et al (2012) Central role of dynamic tidal biofilms dominated by aerobic hydrocarbonoclastic bacteria and diatoms in the biodegradation of hydrocarbons in coastal mudflats. Appl Environ Microbiol 78:3638–3648. https://doi.org/10.1128/AEM.00072-12
De Santana CO, Spealman P, Melo V, Gresham D, de Jesus T, Oliveira E, Chinalia FA (2021) Large-scale differences in diversity and functional adaptations of prokaryotic communities from conserved and anthropogenically impacted mangrove sediments in a tropical estuary. PeerJ 9:e12229. https://doi.org/10.7717/peerj.12229
Diniz C, Cortinhas L, Nerino G, Rodrigues J, Sadeck L, Adami M, Souza-Filho PWM (2019) Brazilian mangrove status: three decades of satellite data analysis. Remote Sens 11:808. https://doi.org/10.3390/rs11070808
Dinsdale EA, Edwards RA, Bailey BA, Tuba I, Akhter S, McNair K et al (2013) Multivariate analysis of functional metagenomes. Front Genet 4:41. https://doi.org/10.3389/fgene.2013.00041
Doherty M, Yager PL, Moran MA, Coles VJ, Fortunato CS, Krusche AV et al (2017) Bacterial biogeography across the Amazon river-ocean continuum. Front Microbiol 8:882. https://doi.org/10.3389/fmicb.2017.00882
Duke NC, Meynecke JO, Dittmann S, Ellison AM, Anger K, Berger U et al (2007) A world without mangroves? Science 317:41–42. https://doi.org/10.1126/science.317.5834.41b
Duran R, Cravo-Laureau C (2016) Role of environmental factors and microorganisms in determining the fate of polycyclic aromatic hydrocarbons in the marine environment. FEMS Microbiol Rev 40:814–830. https://doi.org/10.1093/femsre/fuw031
Falb M, Müller K, Königsmaier L, Oberwinkler T, Horn P, von Gronau S et al (2008) Metabolism of halophilic archaea. Extremophiles 12:177–196. https://doi.org/10.1007/s00792-008-0138-x
Freitas H, Guedes MLS, Smith DH, Oliveira SS, Santos ES, Da Silva EM (2002) Characterization of the mangrove plant community and associated sediment of Todos os Santos Bay, Bahia, Brazil. Aquat Ecosyst Health Manage 5:217–229. https://doi.org/10.1080/14634980290031901
Giri C, Ochieng E, Tieszen LL, Zhu Z, Singh A, Loveland T, Masek J, Duke N (2011) Status and distribution of mangrove forests of the world using earth observation satellite data. Glob Ecol Biogeogr 20:154–159. https://doi.org/10.1111/j.1466-8238.2010.00584.x
Grossart HP, Riemann L, Azam F (2001) Bacterial motility in the sea and its ecological Implications. Aquat Microb Ecol 25:247–258. https://doi.org/10.3354/ame025247
Gutierrez T, Green DH, Nichols PD, Whitman WB, Semple KT, Aitken MD (2013) Polycyclovorans algicola gen. nov., sp. nov., an aromatic-hydrocarbon-degrading marine bacterium found associated with laboratory cultures of marine phytoplankton. Appl Environ Microbiol 79:205–214. https://doi.org/10.1128/AEM.02833-12
Hamilton SE, Casey D (2016) Creation of a high spatio-temporal resolution global database of continuous mangrove forest cover for the 21st century (CGMFC-21). Glob Ecol Biogeogr 25:729–738. https://doi.org/10.1111/geb.12449
Holguin G, Vazquez P, Bashan Y (2001) The role of sediment microorganisms in the productivity, conservation, and rehabilitation of mangrove ecosystems: an overview. Biol Fertil Soils 33:265–278. https://doi.org/10.1007/s003740000319
Huang Y, Fang T, Wang H, Zhou H (2015) Draft genome sequence of Oleiagrimonas soli 3.5XT, a type species in a newly identified genus, isolated from an oil field in China. Genome Announc 3:e00469-15. https://doi.org/10.1128/genomeA.00469-15
Huergo LF, Rissi DV, Elias AS, Gonçalves MV, Gernet MV, Barreto F, Dahmer GW, Reis RA, Pedrosa FO, Souza EM, Monteiro RA, Baura VA, Balsanelli E, Cruz LM (2018) Influence of ancient anthropogenic activities on the mangrove soil microbiome. Sci Total Environ 645:1–9. https://doi.org/10.1016/j.scitotenv.2018.07.094
Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook et al (2019) eggNOG 5.0: a hierarchical, functionally, and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 47:D309–D314. https://doi.org/10.1093/nar/gky1085
Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinforma 11:119. https://doi.org/10.1186/1471-2105-11-119
IBAMA (2020) Instituto Brasileiro do Meio Ambiente e dos Recursos Naturais Renováveis. Relatório nº 6898984/2020-CGEMA/DIPRO (in Brazilian Portuguese). https://www.gov.br/ibama/pt-br/assuntos/fiscalizacao-e-protecao-ambiental/emergencias-ambientais/manchasdeoleo/arquivos/2022/2022-12-16_sei_ibama_6898984_relatorio_tecnico_ibama.pdf. Accessed 15 April 2023
IBGE (2019) Estimativas 2019 população Regiões Metropolitanas. Agência de Notícias - IBGE (in Brazilian Portuguese). https://agenciadenoticias.ibge.gov.br/agencia-detalhe-de-midia.html?view=mediaibge&catid=2103&id=3109. Accessed 10 April 2023
Jansson JK, Hofmockel KS (2018) The soil microbiome—from metagenomics to metaphenomics. Curr Opin Microbiol 43:162–168. https://doi.org/10.1016/j.mib.2018.01.013
Jin M, Guo X, Zhang R, Qu W, Gao B, Zeng R (2019) Diversities and potential biogeochemical impacts of mangrove soil viruses. Microbiome 7:58. https://doi.org/10.1186/s40168-019-0675-9
Karthikeyan S, Rodriguez-R LM, Heritier-Robbins P, Kim M, Overholt WA, Gaby JC et al (2019) “Candidatus Macondimonas diazotrophica”, a novel gammaproteobacterial genus dominating crude-oil-contaminated coastal sediments. ISME J 13:2129–2134. https://doi.org/10.1038/s41396-019-0400-5
Kim KK, Lee KC, Lee JS (2011) Halogranum salarium sp. nov., a halophilic archaeon isolated from sea salt. Syst Appl Microbiol 34:576–580. https://doi.org/10.1016/j.syapm.2011.03.007
Lai J, Cheah W, Palaniveloo K, Suwa R, Sharma S (2022) A systematic review of the physicochemical and microbial diversity of well-preserved, restored, and disturbed mangrove forests: what is known and what is the way forward? Forests 13:2160. https://doi.org/10.3390/f13122160
Leao Z, Dominguez JML (2000) Tropical coast of Brazil. Mar Pollut Bull 41:112–122. https://doi.org/10.1016/S0025-326X(00)00105-3
Lelchat F, Mocaer PY, Ojima T, Michel G, Sarthou G, Bucciarelli E, Cérantola S, Colliec-Jouault S, Boisset C, Baudoux AC (2019) Viral degradation of marine bacterial exopolysaccharides. FEMS Microbiol Ecol 95(7):fiz079. https://doi.org/10.1093/femsec/fiz079
Li Y, Zheng L, Zhang Liu H, Jing H (2019) Comparative metagenomics study reveals pollution induced changes of microbial genes in mangrove sediments. Sci Rep 9:5739. https://doi.org/10.1038/s41598-019-42260-4
Liao S, Wang Y, Liu H, Fan G, Sahu SK, Jin T et al (2020) Deciphering the microbial taxonomy and functionality of two diverse mangrove ecosystems and their potential abilities to produce bioactive compounds. mSystems 5:e00851-19. https://doi.org/10.1128/mSystems.00851-19
Lin H, Peddada SD (2020) Analysis of compositions of microbiomes with bias correction. Nat Commun 11:3514. https://doi.org/10.1038/s41467-020-17041-7
Mai Z, Ye M, Wang Y, Foong SY, Wang L, Sun F, Cheng H (2021) Characteristics of microbial community and function with the succession of mangroves. Front Microbiol 12:764974. https://doi.org/10.3389/fmicb.2021.764974
McMurdie PJ, Holmes S (2013) Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. Plos One 8:e61217. https://doi.org/10.1371/journal.pone.0061217
Mendes LW, Tsai SM (2018) Distinct taxonomic and functional composition of soil microbiomes along the gradient forest-restinga-mangrove in southeastern Brazil. Antonie Van Leeuwenhoek 111(1):101–114. https://doi.org/10.1007/s10482-017-0931-6
Menze BH, Kelm BM, Masuch R, Himmelreich U, Bachert P, Petrich W, Hamprecht FA (2009) A comparison of random forest and its Gini importance with standard chemometric methods for the feature selection and classification of spectral data. BMC Bioinforma 10:213. https://doi.org/10.1186/1471-2105-10-213
Menzel P, Ng KL, Krogh A (2016) Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat Commun 7:11257. https://doi.org/10.1038/ncomms11257
Mori K, Nurcahyanto DA, Kawasaki H, Lisdiyanti P, Yopi, Suzuki KI (2016) Haloarchaeobius baliensis sp. nov., isolated from a solar saltern. Int J Syst Evol Microbiol 66:38–43. https://doi.org/10.1099/ijsem.0.000672
Morris JJ, Rose AL, Lu Z (2022) Reactive oxygen species in the world ocean and their impacts on marine ecosystems. Redox Biol 52:102285. https://doi.org/10.1016/j.redox.2022.102285
Munoz B, Albores A (2011) DNA damage caused by polycyclic aromatic hydrocarbons: mechanisms and markers. In: Chen CC (ed) Selected topics in DNA repair. San Diego, pp 125–144. https://doi.org/10.5772/22527
Nikolaou A, Kostopoulou M, Petsas A, Vagi M, Lofrano G, Meric S (2009) Levels and toxicity of polycyclic aromatic hydrocarbons in marine sediments. TrAC Trends Anal Chem 28:653–664. https://doi.org/10.1016/j.trac.2009.04.004
Nóbrega MS, Silva BS, Tschoeke DA, Appolinario LR, Calegario G, Venas TM, Macedo L, Asp N, Cherene B, Marques JSJ, Seidel M, Dittmar T, Santos IR, de Rezende CE, Thompson CC, Thompson FL (2022) Mangrove microbiome reveals importance of sulfur metabolism in tropical coastal waters. Sci Total Environ 813:151889. https://doi.org/10.1016/j.scitotenv.2021.151889
Nogueira VLR, Rocha LL, Colares GB, Angelim AL, Normando LRO, Cantão ME et al (2015) Microbiomes and potential metabolic pathways of pristine and anthropized Brazilian mangroves. Reg Stud Mar Sci 2:56–64. https://doi.org/10.1016/j.rsma.2015.08.008
Nurk S, Meleshko D, Korobeynikov A, Pevzner PA (2017) metaSPAdes: a new versatile metagenomic assembler. Genome Res 27:824–834. https://doi.org/10.1101/gr.213959.116
Oksanen J, Simpson G, Blanchet F, Kindt R, Legendre P, Minchin P et al (2022) Vegan: community ecology package. R package version 2.6–2, https://CRAN.R-project.org/package=vegan
Paes EDS, Gloaguen TV, Silva HDADC, Duarte TS, de Almeida MDC, Costa ODV, Bomfim MR, Santos JAG (2022) Widespread microplastic pollution in mangrove soils of Todos os Santos Bay, northern Brazil. Environ Res 210:112952. https://doi.org/10.1016/j.envres.2022.112952
Paixão JF, de Oliveira OM, Dominguez JM, dos S Almeida E, Carvalho GC, Magalhães WF (2011) Integrated assessment of mangrove sediments in the Camamu Bay (Bahia, Brazil). Ecotoxicol Environ Saf 74:403–415. https://doi.org/10.1016/j.ecoenv.2010.10.038
Palit K, Rath S, Chatterjee S, Das S (2022) Microbial diversity and ecological interactions of microorganisms in the mangrove ecosystem: threats, vulnerability, and adaptations. Environ Sci Pollut Res 29:32467–32512. https://doi.org/10.1007/s11356-022-19048-7
Pikuta EV, Lyu Z, Hoover RB, Liu Y, Patel NB, Busse HJ, Lawson PA (2017) Williamwhitmania taraxaci gen. nov., sp. nov., a proteolytic anaerobe with a novel type of cytology from Lake Untersee in Antarctica, description of Williamwhitmaniaceae fam. nov., and emendation of the order Bacteroidales Krieg 2012. Int J Syst Evol Microbiol 67:4132–4145. https://doi.org/10.1099/ijsem.0.002266
Punia A, Bharti R (2023) Loss of soil organic matter in the mining landscape and its implication to climate change. Arab J Geosci 16:86. https://doi.org/10.1007/s12517-023-11177-8
R Core Team (2021) R: a language and environment for statistical computing. R foundation for statistical computing, Vienna, Austria. https://www.R-project.org/
Rocha TS, Sales EA, Beretta M, Oliveira IB (2016a) Effects of dredging at Aratu port in All Saints Bay, Brazil: monitoring the metal content in water and sediments. Environ Monit Assess 188:394. https://doi.org/10.1007/s10661-016-5396-y
Rocha LL, Colares GB, Nogueira VLR, Paes FA, Melo VMM (2016b) Distinct habitats select particular bacterial communities in mangrove sediments. Int J Microbiol 2016:3435809. https://doi.org/10.1155/2016/3435809
Sanders CJ, Eyre BD, Santos IR, Machado W, Luiz-Silva W, Smoak JM et al (2014) Elevated rates of organic carbon, nitrogen and phosphorus accumulation in a highly impacted mangrove wetland. Geophys Res Lett 41:2475–2480. https://doi.org/10.1002/2014GL059789
Sczyrba A, Hofmann P, Belmann P, Koslicki D, Janssen S, Dröge J, Gregor I, Majda S, Fiedler J, Dahms E, Bremges A, Fritz A, Garrido-Oter R, Jørgensen TS, Shapiro N, Blood PD, Gurevich A, Bai Y, Turaev D, DeMaere MZ, Chikhi R, Nagarajan N, Quince C, Meyer F, Balvočiūtė M, Hansen LH, Sørensen SJ, Chia BKH, Denis B, Froula JL, Wang Z, Egan R, Don Kang D, Cook JJ, Deltel C, Beckstette M, Lemaitre C, Peterlongo P, Rizk G, Lavenier D, Wu YW, Singer SW, Jain C, Strous M, Klingenberg H, Meinicke P, Barton MD, Lingner T, Lin HH, Liao YC, Silva GGZ, Cuevas DA, Edwards RA, Saha S, Piro VC, Renard BY, Pop M, Klenk HP, Göker M, Kyrpides NC, Woyke T, Vorholt JA, Schulze-Lefert P, Rubin EM, Darling AE, Rattei T, McHardy AC (2017) Critical Assessment of Metagenome Interpretation-a benchmark of metagenomics software. Nat Methods 14(11):1063–1071. https://doi.org/10.1038/nmeth.4458
Silva CS, Moreira IT, de Oliveira OM, Queiroz AF, Garcia KS, Falcão BA, Escobar NF, Rios MC (2014) Spatial distribution and concentration assessment of total petroleum hydrocarbons in the intertidal zone surface sediment of Todos os Santos Bay. Brazil Environ Monit Assess 186:1271–1280. https://doi.org/10.1007/s10661-013-3456-0
Silva LSC, Picanço JL, Calil JGS (2021) O grande desastre esquecido: análise preliminar do derramamento de óleo na costa brasileira (agosto/2019 – março/2020) e seus impactos no litoral da Bahia (in Brazilian Portuguese). Rev Univ Fed Minas Gerais, Belo Horizonte, 27:54–79. https://doi.org/10.35699/2316-770X.2020.21450. https://periodicos.ufmg.br/index.php/revistadaufmg/article/view/21450. Accessed 15 April 2023
Spalding M, Kainuma M, Collins L (2010) In: Earthscan (ed) World atlas of mangroves (version 3.1). A collaborative project of ITTO, ISME, FAO, UNEP-WCMC, UNESCO-MAB, UNU-INWEH, TNC. London, p 319. https://doi.org/10.34892/w2ew-m835
Taketani RG, Yoshiura CA, Dias AC, Andreote FD, Tsai SM (2010) Diversity and identification of methanogenic archaea and sulphate-reducing bacteria in sediments from a pristine tropical mangrove. Antonie Van Leeuwenhoek 97:401–411. https://doi.org/10.1007/s10482-010-9422-8
Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36. https://doi.org/10.1093/nar/28.1.33
Tavares TCL, Bezerra WM, Normando LRO, Rosado AS, Melo VMM (2021) Brazilian semi-arid mangroves-associated microbiome as pools of richness and complexity in a changing world. Front Microbiol 12:715991. https://doi.org/10.3389/fmicb.2021.715991
Thatoi H, Behera BC, Mishra RR, Dutta SK (2013) Biodiversity and biotechnological potential of microorganisms from mangrove ecosystems: a review. Ann Microbiol 63:1–19. https://doi.org/10.1007/s13213-012-0442-7
Valiela I, Bowen JL, York JK (2001) Mangrove forests: one of the world’s threatened major tropical environments. Bioscience 51:807–815. https://doi.org/10.1641/0006-3568(2001)051[0807:MFOOTW]2.0.CO;2
Wang YX, Liu JH, Zhang XX, Chen YG, Wang ZG, Chen Y et al (2009) Fodinicurvata sediminis gen. nov., sp. nov. and Fodinicurvata fenggangensis sp. nov., poly-beta-hydroxybutyrate-producing bacteria in the family Rhodospirillaceae. Int J Syst Evol Microbiol 59:2575–2581. https://doi.org/10.1099/ijs.0.009340-0
Ye SH, Siddle KJ, Park DJ, Sabeti PC (2019) Benchmarking metagenomics tools for taxonomic classification. Cell 178(4):779–794. https://doi.org/10.1016/j.cell.2019.07.010
Acknowledgements
The authors acknowledge the National Laboratory for Scientific Computing (LNCC/MCTI, Brazil) for providing HPC resources of the SDumont supercomputer, contributing to the research results reported in this paper. URL: http://sdumont.lncc.br.
Funding
This study was supported by Petrobras (Process number: 2018/00190–8). Termo de cooperação no. 5900.0109896.18.9/SAP: 4600579545—Sequenciamento de DNA e análises bioinformáticas para metagenômica. ATRV is supported by FAPERJ (E-26/201.046/2022 and E-26/210.012/2020) and CNPq (307145/2021–2).
Author information
Authors and Affiliations
Contributions
J.E.S.P., M.A., C.R.J., and A.T.R.V. conceived the work and designed the experiments. J.E.S.P., M.A., C.R.J. performed the sampling collection. A.L.G and A.P.C.G. performed the metagenomic sequencing. A.T.R.V., L.P.C., F.M.C., and M.L. conceived the work metodology. C.R.J., J.E.S.P., M.A., A.T.R.V., L.C.P., A.L.G., A.P.C.G., F.M.C., M.L, and V.P.K done the data curation. F.M.C and M.L. performed the analyses and interpretation of results. F.M., M.L, and L.P.C wrote the paper, and A.T.R.V., C.R.J., M.A., and J.E.S.P. revised it. C.R.J., J.E.S.P., M.A., A.T.R.V., and L.C.P. supervised the work. All the authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
de Carvalho, F.M., Laux, M., Ciapina, L.P. et al. Finding microbial composition and biological processes as predictive signature to access the ongoing status of mangrove preservation. Int Microbiol (2024). https://doi.org/10.1007/s10123-024-00492-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10123-024-00492-z