
Abstract
Background
Polymyxin B is considered a last-line therapeutic option against multidrug-resistant gram-negative bacteria, especially in COVID-19 coinfections or other serious infections. However, the risk of antimicrobial resistance and its spread to the environment should be brought to the forefront.
Methods
Pandoraea pnomenusa M202 was isolated under selection with 8 mg/L polymyxin B from hospital sewage and then was sequenced by the PacBio RS II and Illumina HiSeq 4000 platforms. Mating experiments were performed to evaluate the transfer of the major facilitator superfamily (MFS) transporter in genomic islands (GIs) to Escherichia coli 25DN. The recombinant E. coli strain Mrc-3 harboring MFS transporter encoding gene FKQ53_RS21695 was also constructed. The influence of efflux pump inhibitors (EPIs) on MICs was determined. The mechanism of polymyxin B excretion mediated by FKQ53_RS21695 was investigated by Discovery Studio 2.0 based on homology modeling.
Results
The MIC of polymyxin B for the multidrug-resistant bacterial strain P. pnomenusa M202, isolated from hospital sewage, was 96 mg/L. GI-M202a, harboring an MFS transporter-encoding gene and conjugative transfer protein-encoding genes of the type IV secretion system, was identified in P. pnomenusa M202. The mating experiment between M202 and E. coli 25DN reflected the transferability of polymyxin B resistance via GI-M202a. EPI and heterogeneous expression assays also suggested that the MFS transporter gene FKQ53_RS21695 in GI-M202a was responsible for polymyxin B resistance. Molecular docking revealed that the polymyxin B fatty acyl group inserts into the hydrophobic region of the transmembrane core with Pi-alkyl and unfavorable bump interactions, and then polymyxin B rotates around Tyr43 to externally display the peptide group during the efflux process, accompanied by an inward-to-outward conformational change in the MFS transporter. Additionally, verapamil and CCCP exhibited significant inhibition via competition for binding sites.
Conclusions
These findings demonstrated that GI-M202a along with the MFS transporter FKQ53_RS21695 in P. pnomenusa M202 could mediate the transmission of polymyxin B resistance.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The relatively rapid global spread of and rise in COVID-19 cases prompted the WHO to declare the disease a pandemic on 11 March 2020 (O'Toole 2021). Studies have indicated that antibiotics are prescribed frequently to patients with COVID-19, largely due to suspected bacterial coinfections (Langford et al. 2021). Despite frequent antibiotic prescription, the prevalence of bacterial coinfection and secondary infection in patients hospitalized with COVID-19 is 3.5% and 14.3%, respectively (Langford et al. 2020). Another major concern regarding hospitalized patients with COVID-19 is bacterial superinfections, especially in the intensive care setting and in patients using invasive devices, as well as in outbreaks of multidrug-resistant bacteria resulting from poorer adherence to infection control practices (Perez et al. 2020; Rawson et al. 2020; Sharifipour et al. 2020). The high frequency of antibiotic prescription highlights the potential for significant antibiotic overuse in these patients (Langford et al. 2021) and antimicrobial resistance (AMR) as a potential consequence. The increase in unnecessary antimicrobial use will potentially enhance the future risk of AMR by driving the selection of multidrug-resistant (MDR) organisms (Rawson et al. 2020). Multiple studies worldwide have reported an unexpectedly high incidence of infections due to methicillin-resistant Staphylococcus aureus (MRSA), methicillin-resistant Acinetobacter baumannii (MRAB), carbapenem-resistant A. baumannii (CRAB), and carbapenem-resistant Enterobacteriaceae (CRE) among COVID-19 patients admitted to the intensive care unit (Segala et al. 2021).
Polymyxins are amphipathic lipopeptide molecules with strong bactericidal activity against a range of gram-negative bacteria. Polymyxin antibiotics, polymyxin B in particular, are increasingly being used as last-line therapeutic options against MDR bacterial infections (Moffatt et al. 2019). Recently, polymyxin B was reported as an effective drug for use in COVID-19 patients with bacterial coinfections (Elmorsy et al. 2022). However, with the increased use of polymyxin B, resistant strains are emerging at an alarming rate. Thus, the repercussions of COVID-19 with bacterial coinfections on AMR are of great concern due to elevated antibiotic use in patients infected with COVID-19 (Hughes et al. 2020). Opportunistic pathogens can also cause superinfections, especially in combination with viral respiratory tract infections in hospitalized patients (Sharifipour et al. 2020). MDR organisms have emerged not only in the hospital environment but also in community settings, suggesting that reservoirs of antibiotic-resistant bacteria are present around hospitals (Youlden et al. 2022). Pandoraea species is a newly emerging multidrug-resistant pathogen usually isolated from a variety of clinical samples (See-Too et al. 2019; Gawalkar et al. 2021; Singh et al. 2021; Cubides-Diaz et al. 2022; Ramos Oliveira et al. 2023) with antibiotic resistance (Schneider et al. 2006). In this study, we isolated the MDR gram-negative strain P. pnomenusa M202 from hospital sewage. M202 could spread polymyxin B resistance via a newly identified MFS transporter present on a genomic island (GI). These results provide new insights into the mechanisms of dissemination of polymyxin B resistance and raise concerns regarding antibiotic therapy for bacterial infections.
Materials and methods
Isolation and identification of the multidrug-resistant P. pnomenusa M202
Hospital sewage was obtained from the wastewater treatment facility in Shandong Province, China. The sewage samples were diluted and spread onto Luria–Bertani (LB) agar plates (0.5% yeast extract, 1% tryptone, 1% sodium chloride, 2% agar) containing 8 mg/L polymyxin B (Sigma Co. Shanghai, China) and then incubated at 28 °C for 24 h. All colonies with different phenotypes on plates were selected and cultivated three consecutive times on LB agar medium with antibiotics to obtain single colonies. After further purification, one single colony, named M202, was selected and grown as a pure culture. For multidrug resistance analysis, antimicrobial susceptibility tests were performed to determine the MICs for 9 antibiotics, including ampicillin, cefixime, tetracycline, ciprofloxacin, florfenicol, amikacin, polymyxin B, sulfamethoxazole, and meropenem, based on the breakpoints defined by the Clinical and Laboratory Standards Institute (CLSI 2020).
Genomic DNA of P. pnomenusa M202 was extracted using the Genomic DNA Purification Kit (Promega, WI, USA) and analyzed using a NanoDrop UV–Vis spectrophotometer (Thermo Scientific, MA, USA). 16S rRNA genes were amplified with the universal primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-TACCTTGTTACGACTT-3′) and sequenced. Similarity analyses of the 16S rRNA sequences were conducted for preliminary identification using BLASTn (https://blast.ncbi.nlm.nih.gov/Blast.cgi). A phylogenetic tree of M202 was produced by using neighbor-joining algorithms in Molecular Evolutionary Genetics Analysis 7 (MEGA 7) software based on the genome sequencing results of the 16S rDNA sequence (Jeong et al. 2016).
Whole-genome sequencing, annotation, and analysis
P. pnomenusa M202 genome was sequenced by the Nanopore and BGISEQ-500 platforms at BGI Co., Ltd. (Wuhan, China). The M202 genome was assembled by glimmer3 (http://www.cbcb.umd.edu/software/glimmer/) with hidden Markov models (Tatusova et al. 2016). Genome annotation was performed using the Prokaryotic Genome Annotation Pipeline of NCBI (http://ncbi.nlm.nih.gov/genome/annotation_prok/). The genome alignment was performed by the PATRIC server (Brettin et al. 2015). GIs were predicted by IslandViewer 4 (Bertelli et al. 2017). The conjugation system was identified by the PATRIC server (Brettin et al. 2015) and oriTfinder (Li et al. 2018).
Analysis of antibiotic resistance genes
Antibiotic resistance genes (ARGs) were analyzed by RAST and BLASTp based on the core dataset in the Antibiotic Resistance Genes Database (ARDB) (Liu and Pop 2009). Multisequence comparison was carried out by Clustal Omega (Madeira et al. 2019) and ESPript (Robert and Gouet 2014).
Mating experiments
Broth-based mating experiments were carried out using M202 as the donor and Escherichia coli 25DN as the recipient as described previously (Zhang et al. 2020; Zong et al. 2020). Transconjugants were screened when 8 mg/L polymyxin B was used for selection. To determine whether GI-M202a was transferred to transconjugants, the transconjugant clone M202-TC1 was analyzed by PCR with three primer pairs (Table S1), including V1-F/R (for the helix-turn-helix domain-containing protein gene, FKQ53_RS21685), V2-F/R (for the MFS gene, FKQ53_RS21695), and V3-F/R (for the RidA family protein gene, FKQ53_RS21700). The MICs of antibiotics for the transconjugants were determined as described above.
Construction of the FKQ53_RS21695 recombinant E. coli strain
The MFS transporter-encoding gene FKQ53_RS21695 in GI-M202a was amplified using the primer pair MRC-F/R (with Xba I at the 5′-end and Hind III at the 3′-end) and M202 genomic DNA as a template. The promoter of the β-lactamase gene was amplified using the primers AP-F/R (with Hind III at the 5′-end and Xba I at the 3′-end) (Table S1) and the pMD18-T vector as a template, and then the above two gene fragments were fused by T4 DNA ligase after Hind III digestion. After digestion by Xba I, the fused fragment was cloned into the pMD18-T vector to obtain recombinant pMD18-Mrc. After verification by DNA sequencing, pMD18-Mrc was transformed into E. coli DH5α (TSINGKE, China) for heterogeneous expression of FKQ53_RS21695, and the recombinant strain was named Mrc-3.
Real-time quantitative PCR analysis
Total RNA isolation and RT-qPCR were performed as described previously (Fu et al. 2017). P. pnomenusa M202, E. coli M202-TC1, and E. coli Mrc-3 were harvested from broth medium at 24 h in the presence of 16 mg/L polymyxin B, and then rapidly frozen in liquid nitrogen. Total RNA was extracted using an RNA extraction kit (SBSBIO, Beijing, China), and then treated with Turbo DNA-free reagents (ABI Ambion, Austin, TX, USA). RT-qPCR assays were performed on a Roche Light Cycler 480 instrument using SYBR Green Mix (Toyobo, Osaka, Japan). Relative quantities of cDNA were normalized to the amounts of the 16S rRNA genes. The primers used are listed in Table S1.
Determination of the MICs with an efflux pump inhibitor
The MICs of polymyxin B for the Mrc-3 strain were calculated by the broth microdilution procedure as described above, and then the MIC of polymyxin B in the presence of efflux pump inhibitors (EPIs) was also calculated. Verapamil, carbonyl cyanide (m-chlorophenyl) hydrazone (CCCP), dihydrochloride (PAβN), and reserpine (RES) were used as EPIs at final concentrations of 8.0 mg/L, 0.1 mg/L, 8.0 mg/L, and 8.0 mg/L, respectively.
Molecular docking of FKQ53_RS21695 with polymyxin B
Transmembrane (TM) regions were identified by TMHMM 2.0 (https://services.healthtech.dtu.dk/service.php? TMHMM-2.0). Protein homology modeling and molecular docking were performed as previously described (Zong et al. 2020). The homology model constructed for the selected MFS transporter was analyzed using Rosetta software (Leman et al. 2020) and Discovery Studio (Biovia 2017). Hydrophobic surface features were analyzed by Discovery Studio (Biovia 2017). The structure of polymyxin B, as a ligand, was obtained from Chemspider (http://www.chemspider.com/). The binding of FKQ53_RS21695 with polymyxin B was modeled using the CDOCKER protocol of Discovery Studio 2.0 (Biovia 2017). The chemical bonds between FKQ53_RS21695 and polymyxin B were demonstrated by 3D and 2D methods.
Results
Polymyxin B resistance of the multidrug-resistant P. pnomenusa M202
P. pnomenusa M202 was isolated from hospital sewage under 8 mg/L polymyxin B selection pressure. MIC analysis of nine antibiotics, including a series of β-lactam, fluoroquinolone, tetracycline, chloramphenicol, aminoglycoside, sulfonamide, carbapenem, and glycopeptide antibiotics, was performed, and the results revealed that M202 was an MDR strain (Table S2). Interestingly, M202 showed high resistance to polymyxin B (MIC: 96 mg/L), which is the last-line therapeutic option against MDR bacteria associated with COVID-19 coinfections or other serious infections.
Isolated strain M202 was identified as P. pnomenusa (Fig. 1A), which is an opportunistic pathogen. M202 harbors a 5.39 Mb circular chromosome with 64.79% GC content (Fig. 1B). The complete genome sequence was annotated, and 1165 functional proteins were assigned Enzyme Commission (EC) numbers, 1013 were assigned Gene Ontology (GO) terms, and 910 were mapped to KEGG pathways (Table S3). Subsystem analysis of the PATRIC annotations indicated that M202 contained 51 stress response, defense, and virulence genes and 39 membrane transport genes (Fig. 1C). The presence of twenty-one virulence factors (Table S4) and two type III secretion systems (Table S5) indicated the risk of clinical infection. Three prophage regions, region 1 (incomplete), region 2 (intact), and region 3 (incomplete), were identified (Table S6). These genes involved in the phage lytic cycle (including holin and tail proteins, gp6-like head-tail connector protein) may increase the possibility resistance transfer (Hernández-Mendoza et al. 2022). Additionally, abundant mobile genetic elements (MGEs), including thirty-nine insertion sequences (ISs) with transposases and five integrases (Table S7), showed that the diversity of the M202 genome may be attributed to horizontal gene transfer and that M202 also has the ability to spread antibiotic resistance.

Identification and genomic characteristics of M202. A Neighbor-joining tree generated on the basis of 16S rDNA gene sequences of M202. B PATRIC annotation of the genome of M202. C Subsystem analysis of the M202 genome
Antimicrobial resistance genes and potential antibiotic genomic islands in M202
Genes that are responsible for resistance to β-lactam, carbapenem, and tetracycline, including β-lactamase, AmpC, OXA-62, MBL metallo-hydrolase, and ribosome protection-type encoding genes, could be identified (Table 1). In addition, a considerable number of efflux pump genes were identified, and the major facilitator superfamily (MFS) and ATP-binding cassette (ABC) transporters were prominent classes, with 128 and 277 related genes, respectively (Table 1). In addition, lipopolysaccharide modification gene pmrK, an UDP phosphate-alpha-4-amino-4-deoxy-L-arabinose arabinosyl transferase encoding gene which was reported to be the main mechanism of polymyxin resistance in gram-negative bacteria (Moffatt et al. 2019), was also found in the genome of M202 (Table 1).
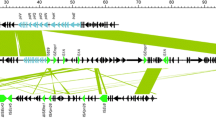
To analyze the transfer and polymyxin resistance mechanism of M202, mobilizable GIs were investigated. Considered that pmrK (from 3,208,569 to 3,210,299 bp in genome) was not located in any GI, it is not the reason for polymyxin B resistance transfer ability. Thus, potential mobilizable GIs harboring antibiotic resistance genes were analyzed. Two overlapping regions, the first ranging from nucleotide positions 4904719 to 4940759 and the second from positions 4924191 to 4934831, were annotated as potential antibiotic GIs by IslandPath-DIMOB and SIGI-HMM, respectively. Further alignment analysis indicated that a single GI most likely extended from position 4904719 to 4942265, and this region was named GI-M202a (Fig. 2). Sequence examination further suggested that integration of GI-M202a may have occurred within the intergenic sequence between FKQ53_RS21670 and FKQ53_RS21875 and that this resulted in the formation of the incomplete attachment (att) sites GCTTTTATA and GCTTTTTTAT, which are similar to attL and attR of phage region 1 (Rutherford et al. 2013). Thirty-seven ORFs were identified in GI-M202a (Fig. 2); among them, four horizontal gene transfer elements, including two transposases (one of the IS3 family and the other of the IS30 family), two type IV conjugative transfer proteins (including a shufflon-specific DNA recombinase), one mobilization relaxosome protein (MobC), and one large-conductance mechanosensitive channel (FKQ53_RS21860), were predicted. In addition, one MFS transporter (FKQ53_RS21695) that might be related to antibiotic resistance was identified.

Identification of GI-M202a in the M202 genome. Top image, GIs predicted by IslandViewer 4. Putative GIs predicted by the SIGI-HMM method (yellow squares) or IslandPath-DIMOB method (blue squares). The integrated results are indicated by red squares. Bottom line, gene arrangement in GI-M202a. Genes are denoted by arrows. MFS transporter, green arrows; DNA transfer-related genes, gray arrows; genes of unknown function, purple arrows; type IV secretion system, orange arrows; others, blue arrows
An MFS transporter (FKQ53_RS21695) is responsible for polymyxin B resistance
PCR and DNA sequencing analysis confirmed that GI-M202a had been transferred to M202-TC1 (Fig. S1), indicating that the transferred gene conferring resistance to polymyxin B was in GI-M202a and was responsible for the polymyxin B resistance of M202-TC1. Considering that GI-M202a harbored one MFS transporter (FKQ53_RS21695), the EPIs CCCP and verapamil were used to evaluate efflux-mediated polymyxin B resistance as described previously (Fu et al. 2020). The MICs of polymyxin B for P. pnomenusa M202 and the E. coli transconjugant M202-TC1 clearly decreased from 96 to 24 mg/L and 24 mg/L in the presence of CCCP and verapamil, while PaβN and reserpine did not affect the MICs (Fig. 3A), suggesting that the MFS transporter (FKQ53_RS21695) in GI-M202a might be associated with polymyxin B resistance transfer.

A MICs of polymyxin B for different strains. M202-TC1: transconjugant of M202 and E. coli 25DN; Mrc-3: DH5α with heterogeneous expression of FKQ53_RS21695. B RT-qPCR analyses of FKQ53_RS21695 gene in P. pnomenusa M202, E. coli M202-TC1, and E. coli Mrc-3. **: p < 0.001
To determine whether the MFS transporter (FKQ53_RS21695) is responsible for the polymyxin B resistance of M202, the encoding gene FKQ53_RS21695 was heterogeneously expressed in E. coli DH5α, and the recombinant strain was named Mrc-3. MIC analysis showed that Mrc-3 acquired resistance to polymyxin B (MIC 16 mg/L), and CCCP or verapamil clearly reduced the MICs of polymyxin B for the Mrc-3 strain to 2.0 mg/L, respectively (Fig. 3A). Real-time quantitative PCR analyses showed that expressions of FKQ53_RS21695 were higher in the presence of polymyxin B than the control group, both in P. pnomenusa M202 (increased by 2.56-fold) and E. coli M202-TC1 (increased 3.22-fold) (Fig. 3B). Moreover, expressions of FKQ53_RS21695 in E. coli Mrc-3 were detected, although it showed no significant difference between with and without polymyxin B. This result indicating that the MFS transporter FKQ53_RS21695 contributes polymyxin B resistance and transfer.
Overall structure of the MFS transporter FKQ53_RS21695
The MFS family protein FKQ53_RS21695 in GI-M202a consists of 450 amino acid residues. Similar to previously reported MFS transporter structures (Li et al. 2015), homologous modeling showed that the crystal structure of FKQ53_RS21695 contains 12 TM helices (TMs 1–12) and forms two domains (C domain and N domain) (Fig. 4A). The two domains are connected by a 34-residue linker (residues 223–246) containing a three-turn amphipathic α-helix as well as an extended loop.

Overall structure of the MFS transporter FKQ53_RS21695 of GI-M202a. A Cartoon representation of the FKQ53_RS21695 structure. The N-domain, C-domain, and α′ are represented in blue, orange, and green, respectively. Numbers indicate the TM helixes. B Active conformation of motif A and protonation/deprotonation-related residues. C The overall hydrophobic surface of FKQ53_RS21695. D The cavity-facing sides of the N and C domains have contrasting surface electrostatic potentials. The figure was generated using DS
Most MFS transporters contain the signature motif A, “G(+1)xlaD(+5) rxGR(+9)kp” (Paulsen et al. 1996; Jiang et al. 2013). However, motif A-1 of FKQ53_RS21695, located in the loop connecting TMs 2 and 3 (L2–3, residues 86–95), was identified as “NMMMHRFGAR”. Similarly, motif A-2 was “RHSDRTMERR” and was located in the loop connecting TMs 8 and 9 (L8–9, residues 314–323) (Fig. 4B). This unusual motif made TM2 distant from TM11 and unable to form a close helix-helix contact, as also observed for E. coli YajR (Jiang et al. 2013). However, Arg95, which was proven to be important to the thermal stability and transport activity of MFS efflux proteins in YajR (Arg77) (Jiang et al. 2013) and TetA (Arg71) (Bannam et al. 2004), was conserved. Unlike the enriched Asp or Glu to be deprotonated during transport (Dang et al. 2010; Jiang et al. 2013; Heng et al. 2015; Leano et al. 2019), FKQ53_RS21695 maintained Asp45 and Glu132, which are located on the third helical turn of the corresponding TM1 and TM4 in the two internal structural repeats of the N domain amphipathic cavity (Fig. 4B). Therefore, only these residues can undergo cycles of protonation and deprotonation along the transport path.
The N and C domains of FKQ53_RS21695 have hydrophobic surface features. The N domain is capped by positively charged residues on the periplasmic side and by negatively charged residues on the cytoplasmic side (Fig. 4C). In addition, FKQ53_RS21695 has a central cavity in the TM core between the N and C domains. The N domain with a slight hydrophilic surface and C domain with a hydrophilic surface give rise to the amphipathic cavity of the TM core (Fig. 4D).
Mechanism of polymyxin B efflux mediated by the MFS transporter FKQ53_RS21695
To analyze the details of polymyxin B efflux mediated by the MFS transporter FKQ53_RS21695, the interactions between polymyxin B and FKQ53_RS21695 were analyzed by molecular simulation. The outward-open and inward-open conformations of FKQ53_RS21695 showed the polymyxin B binding cavities (Fig. 5). In the inward-open conformation, the polymyxin B binding cavity (PBC1) was located in the central cavity in the TM core between the N and C domains. PBC presented a subulate form, which was embedded in the central cavity between the N and C domains (Fig. 5A). Similar to the inward-open conformation, PBC2 also presented subulate forms in the outward-open conformation (Fig. 5B). The cuspidal termini of PBC, in both outward-open and inward-open conformations, were located close to the protonation and deprotonation residues (PDR) (Fig. 5A–B). These results indicated that polymyxin B may “turn around” in the amphipathic cavity of the TM core during the conformational transition of FKQ53_RS21695.

Polymyxin B efflux mediated by the MFS transporter FKQ53_RS21695. A PBC in the inward-open conformation. B PBC in outward-open conformation. PBC, polymyxin B binding cavity; PDR, protonation and deprotonation residues. C Interactions of PBC with polymyxin B in the inward-open conformation. D Interactions of PBC with polymyxin B in the outward-open conformation. Left: location of polymyxin B; right: interacting amino acid residues and chemical bonds
Polymyxin B is an amphipathic lipopeptide molecule that contains a hydrophobic fatty acyl group at the N-terminus and a hydrophilic heptapeptide (Cai et al. 2015). The fatty acyl group at the N-terminus and the side chains of the residues at positions 6 (p6) and 7 (p7) contribute to the apolar portion (Cai et al. 2015). During efflux, the fatty acyl group inserts into the amphipathic cavity of the TM core for inward-open conformation and partially into the hydrophobic region. Phe273, Tyr43, Phe272, and Tyr265 of FKQ53_RS21695 were the main contributors located in binding cavities for fatty acyl group binding by Pi-alkyl and unfavorable bump interactions (Fig. 5C). The peptide fragment of FKQ53_RS21695 was bound by residues of TM10 and TM11 mainly via unfavorable bump interactions (Fig. 5C). This nature of the interaction allowed the peptide fragment to easily escape the binding, leading to a change in the orientation. After protonation and conformational change from inward-open to outward-open, the binding of polymyxin B moved from Phe273, Tyr43, Phe272, and Tyr265 to the adjacent amino acid residues Phe272, Tyr43, Leu167, Ser276, and Ile275. Moreover, the unfavorable bump interactions changed to the main chemical bonds binding FKQ53_RS21695 and polymyxin B, both the fatty acyl group and peptide fragment (Fig. 5D). Thus, separation from the outward-open conformation of FKQ53_RS21695 became easier than that from the inward-open conformation.
Moreover, FKQ53_RS21695 showed binding with verapamil and CCCP. Interestingly, these binding cavities for EPIs in FKQ53_RS21695 partly overlapped with the polymyxin B binding cavity (Fig. 6). This indicated that the inhibitory effect of verapamil and CCCP on polymyxin B resistance may be attributed to binding site competition. The key EPI-binding amino acids were Phe136, Arg46, Tyr160, and Phe272/273 (Fig. S2). The polymyxin B efflux-related key amino acids, Phe272 and Phe273, bind to verapamil and CCCP, leading to attenuation of polymyxin B binding and “turn around” movement in the amphipathic cavity of FKQ53_RS21695.

Binding cavities for verapamil and CCCP in FKQ53_RS21695. A Locations of the polymyxin B- and verapamil-binding cavities. B Locations of the polymyxin B- and CCCP-binding cavities
Discussion
Gram-negative microorganisms are associated with coinfections in patients with COVID-19 (Povoa et al. 2020; Sehgal et al. 2021; Singh et al. 2021; Cubides-Diaz et al. 2022). Pandoraea species, which are gram-negative bacteria, can cause bacteremia and lung function decline (Lin et al. 2019). P. pnomenusa superinfection in a patient with COVID-19 pneumonia was found in 2022 in Colombia (Cubides-Diaz et al. 2022). Pandoraea spp. are resistant to many antibiotics, which makes the treatment of Pandoraea-related infections more complicated (Lin et al. 2019). Polymyxin B is thought to be a last-line therapeutic option against MDR gram-negative bacteria (Katagiri et al. 2022) and has been used to treat coinfections in patients with COVID-19. However, we isolated P. pnomenusa strain M202 with polymyxin B resistance from hospital sewage, which caught our attention. Hence, exploration of resistance to the emergency antibiotic polymyxin B, after 3 years of the pandemic, is urgently needed.
Moreover, P. pnomenusa M202 is also resistant to ampicillin, cefixime, tetracycline, amikacin, sulfamethoxazole, and meropenem. This multidrug-resistant strain contains several antibiotic resistance genes, such as β-lactamase encoding genes, Tcr (tetracycline-resistant gene), and efflux pump genes. Especially, carbapenem-hydrolyzing oxacillinase OXA-62, which was found to be a novel mechanism of carbapenem resistance in P. pnomenusa (Schneider et al. 2006), was also identified in M202 genome (Table 1). In addition, multidrug-resistant P. pnomenusa strains RB-44, which was reported at 2014 in Malaysia (Ee et al. 2014), also harbored plenty of antibiotic resistance genes, including class A, B, C, and D β-lactamase encoding genes, tetracycline-resistant genes, and efflux pumps genes (Table 1). These data indicate a potential reservoir of antibiotic resistance genes in this species and a further complication in the treatment of infections caused by P. pnomenusa.
Polymyxins have been used to treat MDR gram-negative bacterial infections. It is generally accepted that gram-negative selectivity is mediated by initial interactions with the outer membrane with negatively charged lipopolysaccharides (LPSs), where the cationic polypeptide portion of polymyxin electrostatically binds. LPSs are the predominant (or only) surface lipids of the outer membrane in gram-negative bacteria, and the binding is assisted by the interactions of the lipid tail with the fatty acids of the lipid A moiety of the LPS molecules (Fernandez et al. 2013; Trimble et al. 2016). The polymyxin resistance mechanism mainly involves alterations to reduce the net negative charge or fluidity of LPS (summarized in Trimble et al. 2016). However, despite the membrane being the sole target for polymyxins, alternative or additional mechanisms of action likely contribute to the antibacterial activity of polymyxins. Intracellular components such as the 16S A-site of E. coli ribosomes (McCoy et al. 2013) and the oxidative stress response gene soxS (Dong et al. 2015) are potential targets. However, polymyxin-resistant pathogens pose a serious threat to human health. In this study, the mobilizable antibiotic resistance island GI-M202a was found and identified as a novel antibiotic resistance island that confers polymyxin B resistance. This enhanced risk of polymyxin B resistance spreading to the environment needs to be monitored closely, especially in the context of COVID-19 coinfections, before a better therapeutic regimen is developed.
MFS transporters constitute the largest class of secondary transporters and share a similar folding topology and function (Huang et al. 2003; Jiang et al. 2013). MFS proteins are believed to switch between two major conformations, inward and outward, which differ by an ∼40° rotation of one domain relative to the other (Jiang et al. 2013). The conformational changes can be further regulated by substrate binding inside the cavity or at allosteric sites on either side of the TM core, depending on the direction of substrate transport (Guan and Kaback 2004; Jiang et al. 2013). Most MFS proteins are located in the IM and transport drugs from the cytosol to the periplasm (Li et al. 2015). Similar to the typical MFS structure, FKQ53_RS21695 contains a 12 TM helix core composed of two six-helix rigid domains forming a central TM channel and two antibiotic binding cavities (each present in each conformation) with different interactions with and affinities for antibiotics, similar to ABC transporters coupled with ATP hydrolysis (Orelle et al. 2003; Hofmann et al. 2019). The pathway by which polymyxin B is discharged from the periplasm to the extracellular space may need the assistance of a membrane fusion protein or porin, similar to the EmrAB-TolC system (Tanabe et al. 2009; Blair et al. 2014), resistance-nodulation-division transporter ArcAB-TolC system (Nolivos et al. 2019), or HlyBD-TolC system (Holland et al. 2016). In this study, we found that the MFS transporter FKQ53_RS21695 could bind and facilitate efflux of polymyxin B. During the efflux process, the polymyxin B fatty acyl group inserts into the hydrophobic region of the TM core with Pi-alkyl and unfavorable bump interactions. Then, polymyxin B turns over around Tyr43 to externally display the peptide group, accompanied by the generation of more unfavorable bumps in the outward conformation. Finally, the mechanism by which polymyxin B passes through the FKQ53_RS21695 TM core is proposed (Fig. 7). In addition, a previous study revealed that insertional inactivation of the MFS gene kpnGH resulted in increased susceptibility to polymyxin-B in Klebsiella pneumoniae (Srinivasan et al. 2014), also indicating the polymyxin B function of the MFS transporter. However, this antibiotic transport mode of antibiotic binding cavities associated with proton transport needs further investigation.

Proposed polymyxin B transport mechanism mediated by FKQ53_RS21695. Polymyxin B “turn around” transport mode
Conclusions
The results of this study reflect that the MDR bacterial P. pnomenusa M202 is an effective disseminator of polymyxin B resistance. The MFS efflux pump FKQ53_RS21695 in GI-M202a is responsible for the polymyxin B resistance. FKQ53_RS21695 facilitates polymyxin B efflux through its amphipathic core, which is composed of TM regions. The penetrating power is related to different interactions between MFS transporters and polymyxin B when FKQ53_RS21695 presents inward and outward conformations.
Data availability
The datasets supporting the conclusions of this article are available from the lead authors upon reasonable request.
References
Bannam TL, Johanesen PA, Salvado CL, Pidot SJA, Farrow KA, Rood JI (2004) The Clostridium perfringens TetA(P) efflux protein contains a functional variant of the motif A region found in major facilitator superfamily transport proteins. Microbiology 150:127–134. https://doi.org/10.1099/mic.0.26614-0
Bertelli C, Laird MR, Williams KP, Simon Fraser University Research Computing, G, Lau BY, Hoad G et al (2017) IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res 45:30–35. https://doi.org/10.1093/nar/gkx343
Biovia (2017) Discovery Studio modeling environment, release 2017. Dassault Systemes, San Diego, CA
Blair JM, Richmond GE, Piddock LJ (2014) Multidrug efflux pumps in gram-negative bacteria and their role in antibiotic resistance. Future Microbiol 9:1165–1177. https://doi.org/10.2217/fmb.14.66
Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ et al (2015) RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep 5:8365. https://doi.org/10.1038/srep08365
Cai Y, Lee W, Kwa AL (2015) Polymyxin B versus colistin: an update. Expert Rev Anti Infect Ther 13:1481–1497. https://doi.org/10.1586/14787210.2015.1093933
CLSI (2020) Performance standards for antimicrobial susceptibility testing. Clinical and Laboratory Standards Institute
Cubides-Diaz DA, Munoz Angulo N, Martin Arsanios DA, Ovalle Monroy AL, Perdomo-Rodriguez DR, Del-Portillo MP (2022) Pandoraea pnomenusa superinfection in a patient with SARS-CoV-2 Pneumonia: first case in the literature. Infect Dis Rep 14:205–212. https://doi.org/10.3390/idr14020025
Dang S, Sun L, Huang Y, Lu F, Liu Y, Gong H et al (2010) Structure of a fucose transporter in an outward-open conformation. Nature 467:734–738. https://doi.org/10.1038/nature09406
Dong TG, Dong S, Catalano C, Moore R, Liang X, Mekalanos JJ (2015) Generation of reactive oxygen species by lethal attacks from competing microbes. Proc Natl Acad Sci U S A 112:2181–2186. https://doi.org/10.1073/pnas.1425007112
Ee R, Lim YL, Yin WF, Chan KG (2014) De novo assembly of the quorum-sensing Pandoraea sp. strain RB-44 complete genome sequence using PacBio single-molecule real-time sequencing technology. Genome Announc 2. https://doi.org/10.1128/genomeA.00245-14
Elmorsy MA, El-Baz AM, Mohamed NH, Almeer R, Abdel-Daim MM, Yahya G (2022) In silico screening of potent inhibitors against COVID-19 key targets from a library of FDA-approved drugs. Environ Sci Pollut Res Int 29:12336–12346. https://doi.org/10.1007/s11356-021-16427-4
Fernandez L, Alvarez-Ortega C, Wiegand I, Olivares J, Kocincova D, Lam JS et al (2013) Characterization of the polymyxin B resistome of Pseudomonas aeruginosa. Antimicrob Agents Chemother 57:110–119. https://doi.org/10.1128/AAC.01583-12
Fu J, Zhong C, Zhang P, Zong G, Liu M, Cao G (2020) Novel mobilizable genomic island GEI-D18A mediates conjugational transfer of antibiotic resistance genes in the multidrug-resistant strain Rheinheimera sp. D18. Front Microbiol 11:627. https://doi.org/10.3389/fmicb.2020.00627
Fu JF, Zong G, Zhang P, Zhao Z, Ma J, Pang X, Cao G (2017) XdhR negatively regulates actinorhodin biosynthesis in Streptomyces coelicolor M145. FEMS Microbiol Lett 364. https://doi.org/10.1093/femsle/fnx226
Gawalkar AA, Kasinadhuni G, Kanaujia R, Rajan P, Vijay J, Revaiah PC et al (2021) Prosthetic aortic valve dehiscence following infective endocarditis by a rare bacterium - Pandoraea pnomenusa. J Cardiol Cases 24:27–29. https://doi.org/10.1016/j.jccase.2020.12.003
Guan L, Kaback HR (2004) Binding affinity of lactose permease is not altered by the H+ electrochemical gradient. Proc Natl Acad Sci U S A 101:12148–12152. https://doi.org/10.1073/pnas.0404936101
Heng J, Zhao Y, Liu M, Liu Y, Fan J, Wang X et al (2015) Substrate-bound structure of the E. coli multidrug resistance transporter MdfA. Cell Res 25:1060–1073. https://doi.org/10.1038/cr.2015.94
Hernández-Mendoza A, Salgado-Morales R, Morán-Vázquez A, López-Torres D, García-Gómez BI, Dantán-González E (2022) Molecular characterization of pBOq-IncQ and pBOq-95LK plasmids of Escherichia coli BOq 01, a new isolated strain from poultry farming, involved in antibiotic resistance. Microorganisms 10. https://doi.org/10.3390/microorganisms10081509
Hofmann S, Januliene D, Mehdipour AR, Thomas C, Stefan E, Bruchert S et al (2019) Conformation space of a heterodimeric ABC exporter under turnover conditions. Nature 571:580–583. https://doi.org/10.1038/s41586-019-1391-0
Holland IB, Peherstorfer S, Kanonenberg K, Lenders M, Reimann S, Schmitt L (2016) Type I protein secretion-deceptively simple yet with a wide range of mechanistic variability across the family. EcoSal Plus 7. https://doi.org/10.1128/ecosalplus.ESP-0019-2015
Huang Y, Lemieux MJ, Song J, Auer M, Wang DN (2003) Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 301:616–620. https://doi.org/10.1126/science.1087619
Hughes S, Troise O, Donaldson H, Mughal N, Moore LSP (2020) Bacterial and fungal coinfection among hospitalized patients with COVID-19: a retrospective cohort study in a UK secondary-care setting. Clin Microbiol Infect 26:1395–1399. https://doi.org/10.1016/j.cmi.2020.06.025
Jeong S, Hong JS, Kim JO, Kim K-H, Lee W, Bae IK et al (2016) Identification of Acinetobacter species using matrix-assisted laser desorption ionization-time of flight mass spectrometry. Ann Lab Med 36:325–334. https://doi.org/10.3343/alm.2016.36.4.325
Jiang D, Zhao Y, Wang X, Fan J, Heng J, Liu X et al (2013) Structure of the YajR transporter suggests a transport mechanism based on the conserved motif A. Proc Natl Acad Sci U S A 110:14664–14669. https://doi.org/10.1073/pnas.1308127110
Katagiri D, Izumi S, Takano H (2022) When should polymyxin B-immobilized polystyrene column be introduced to improve COVID-19 prognosis? Ther Apher Dial 26:550–551. https://doi.org/10.1111/1744-9987.13825
Langford BJ, So M, Raybardhan S, Leung V, Soucy J-PR, Westwood D et al (2021) Antibiotic prescribing in patients with COVID-19: rapid review and meta-analysis. Clin Microbiol Infect 27:520–531. https://doi.org/10.1016/j.cmi.2020.12.018
Langford BJ, So M, Raybardhan S, Leung V, Westwood D, MacFadden DR et al (2020) Bacterial co-infection and secondary infection in patients with COVID-19: a living rapid review and meta-analysis. Clin Microbiol Infect 26:1622–1629. https://doi.org/10.1016/j.cmi.2020.07.016
Leano JB, Batarni S, Eriksen J, Juge N, Pak JE, Kimura-Someya T et al (2019) Structures suggest a mechanism for energy coupling by a family of organic anion transporters. PLoS Biol 17:e3000260. https://doi.org/10.1371/journal.pbio.3000260
Leman JK, Weitzner BD, Lewis SM, Adolf-Bryfogle J, Alam N, Alford RF et al (2020) Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat Methods 17:665–680. https://doi.org/10.1038/s41592-020-0848-2
Li X, Xie Y, Liu M, Tai C, Sun J, Deng Z, Ou H-Y (2018) oriTfinder: a web-based tool for the identification of origin of transfers in DNA sequences of bacterial mobile genetic elements. Nucleic Acids Res 46:229–234. https://doi.org/10.1093/nar/gky352
Li XZ, Plesiat P, Nikaido H (2015) The challenge of efflux-mediated antibiotic resistance in gram-negative bacteria. Clin Microbiol Rev 28:337–418. https://doi.org/10.1128/CMR.00117-14
Lin C, Luo N, Xu Q, Zhang J, Cai M, Zheng G, Yang P (2019) Pneumonia due to Pandoraea apista after evacuation of traumatic intracranial hematomas: a case report and literature review. BMC Infect Dis 19:869. https://doi.org/10.1186/s12879-019-4420-6
Liu B, Pop M (2009) ARDB—antibiotic resistance genes database. Nucleic Acids Res 37:443–447. https://doi.org/10.1093/nar/gkn656
Madeira F, Park YM, Buso JLN, Gur T, Madhusoodanan N, Basutka P et al (2019) The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 47:636–641. https://doi.org/10.1093/nar/gkz268
McCoy LS, Roberts KD, Nation RL, Thompson PE, Velkov T, Li J, Tor Y (2013) Polymyxins and analogues bind to ribosomal RNA and interfere with eukaryotic tanslation in vitro. ChemBioChem 14:2083–2086. https://doi.org/10.1002/cbic.201300496
Moffatt JH, Harper M, Boyce JD (2019) Mechanisms of polymyxin resistance. Adv Exp Med Biol 1145:55–71. https://doi.org/10.1007/978-3-030-16373-0_5
Nolivos S, Cayron J, Dedieu A, Page A, Delolme F, Lesterlin C (2019) Role of AcrAB-TolC multidrug efflux pump in drug-resistance acquisition by plasmid transfer. Science 364
Orelle C, Dalmas O, Gros P, Di Pietro A, Jault JM (2003) The conserved glutamate residue adjacent to the Walker-B motif is the catalytic base for ATP hydrolysis in the ATP-binding cassette transporter BmrA. J Biol Chem 278:47002–47008. https://doi.org/10.1074/jbc.M308268200
O'Toole RF (2021) The interface between COVID-19 and bacterial healthcare-associated infections. Clin Microbiol Infect 27:1772–1776. https://doi.org/10.1016/j.cmi.2021.06.001
Paulsen IT, Brown MH, Skurray RA (1996) Proton-dependent multidrug efflux systems. Microbiol Rev 60:575–608. https://doi.org/10.1128/mr.60.4.575-608.1996
Perez S, Innes GK, Walter MS, Mehr J, Arias J, Greeley R, Chew D (2020) Increase in hospital-acquired carbapenem-resistant Acinetobacter baumannii infection and colonization in an acute care-hospital during a surge in COVID-19 admissions. MMWR Morb Mortal Wkly Rep 69:1827–1831. https://doi.org/10.15585/mmwr.mm6948e1
Povoa HCC, Chianca GC, Iorio N (2020) COVID-19: an alert to ventilator-associated bacterial Pneumonia. Infect Dis Ther 9:417–420. https://doi.org/10.1007/s40121-020-00306-5
Ramos Oliveira S, Costa Monteiro I, Rodrigues C, Soares MJ (2023) Fever in a patient with a central venous catheter colonized by Pandoraea pnomenusa. Acta Med Port 36:127–130. https://doi.org/10.20344/amp.16176
Rawson TM, Ming D, Ahmad R, Moore LSP, Holmes AH (2020) Antimicrobial use, drug-resistant infections and COVID-19. Nat Rev Microbiol 18:409–410. https://doi.org/10.1038/s41579-020-0395-y
Robert X, Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:320–324. https://doi.org/10.1093/nar/gku316
Rutherford K, Yuan P, Perry K, Sharp R, Van Duyne GD (2013) Attachment site recognition and regulation of directionality by the serine integrases. Nucleic Acids Res 41:8341–8356. https://doi.org/10.1093/nar/gkt580
Schneider I, Queenan AM, Bauernfeind A (2006) Novel carbapenem-hydrolyzing oxacillinase OXA-62 from Pandoraea pnomenusa. Antimicrob Agents Chemother 50:1330–1335. https://doi.org/10.1128/aac.50.4.1330-1335.2006
See-Too WS, Ambrose M, Malley R, Ee R, Mulcahy E, Manche E et al (2019) Pandoraea fibrosis sp. nov., a novel Pandoraea species isolated from clinical respiratory samples. Int J Syst Evol Microbiol 69:645–651. https://doi.org/10.1099/ijsem.0.003147
Segala FV, Bavaro DF, Di Gennaro F, Salvati F, Marotta C, Saracino A et al (2021) Impact of SARS-CoV-2 epidemic on antimicrobial resistance: a literature review. Viruses 13:2110. https://doi.org/10.3390/v13112110
Sehgal K, Fadel HJ, Tande AJ, Pardi DS, Khanna S (2021) Outcomes in patients with SARS-CoV-2 and Clostridioides difficile coinfection. Infect Drug Resist 14:1645–1648. https://doi.org/10.2147/IDR.S305349
Sharifipour E, Shams S, Esmkhani M, Khodadadi J, Fotouhi-Ardakani R, Koohpaei A et al (2020) Evaluation of bacterial co-infections of the respiratory tract in COVID-19 patients admitted to ICU. BMC Infect Dis 20:646. https://doi.org/10.1186/s12879-020-05374-z
Singh S, Sahu C, Patel SS, Garg A, Ghoshal U (2021) Pandoraea apista bacteremia in a COVID-positive man: a rare coinfection case report from North India. J Lab Physicians 13:192–194. https://doi.org/10.1055/s-0041-1730847
Srinivasan VB, Singh BB, Priyadarshi N, Chauhan NK, Rajamohan G (2014) Role of novel multidrug efflux pump involved in drug resistance in Klebsiella pneumoniae. PLoS One 9:e96288. https://doi.org/10.1371/journal.pone.0096288
Tanabe M, Szakonyi G, Brown KA, Henderson PJ, Nield J, Byrne B (2009) The multidrug resistance efflux complex, EmrAB from Escherichia coli forms a dimer in vitro. Biochem Biophys Res Commun 380:338–342. https://doi.org/10.1016/j.bbrc.2009.01.081
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki E, Pruitt KD et al (2016) NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 44:6614–6624. https://doi.org/10.1093/nar/gkw569
Trimble MJ, Mlynarcik P, Kolar M, Hancock RE (2016) Polymyxin: alternative mechanisms of action and resistance. Cold Spring Harb Perspect Med 6:a025288. https://doi.org/10.1101/cshperspect.a025288
Youlden G, McNeil HE, Blair JMA, Jabbari S, King JR (2022) Mathematical modelling highlights the potential for genetic manipulation as an adjuvant to counter efflux-mediated MDR in Salmonella. Bull Math Biol 84:56. https://doi.org/10.1007/s11538-022-01011-9
Zhang PP, Liu M, Fu J, Zhong C, Zong G, Cao G (2020) Identification of a mobilizable, multidrug-resistant genomic island in Myroides odoratimimus isolated from Tibetan pasture. Sci Total Environ 723:137970. https://doi.org/10.1016/j.scitotenv.2020.137970
Zong G, Zhong C, Fu J, Zhang Y, Zhang P, Zhang W et al (2020) The carbapenem resistance gene blaOXA-23 is disseminated by a conjugative plasmid containing the novel transposon Tn6681 in Acinetobacter johnsonii M19. Antimicrob Resist Infect Control 9:182. https://doi.org/10.1186/s13756-020-00832-4
Acknowledgements
We thank Jennifer Smith, PhD, from Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Funding
This work was supported by the Academic Promotion Program of Shandong First Medical University (grant number LJ001, 2019), the Innovation Project of the Shandong Academy of Medical Sciences (grant number 2022), and the National College Students’ Innovation and Entrepreneurship Training Program (grant number 2029, 2022).
Author information
Authors and Affiliations
Contributions
WHG co-led the data analysis and wrote the manuscript. CCL co-led the data analysis and edited the manuscript. FTW prepared the hospital sewage and supervised data analysis. YLY co-led the data analysis. LZ co-led the bioinformatics analysis. ZXW helped to analyze data. XC helped to analyze data and revised the manuscript. MXT co-led the bioinformatics analysis. GXC co-led the project and revised the manuscript. GLZ co-led the project and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors approved the manuscript.
Competing interests
The authors declare no competing interests.
Nucleotide sequence accession number
Complete sequences of the Pandoraea pnomenusa M202 genome were deposited in GenBank under accession no. CP041237.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOC 1004 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gao, W., Li, C., Wang, F. et al. An efflux pump in genomic island GI-M202a mediates the transfer of polymyxin B resistance in Pandoraea pnomenusa M202. Int Microbiol 27, 277–290 (2024). https://doi.org/10.1007/s10123-023-00384-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10123-023-00384-8