
Abstract
Background
The PI3K/AKT signaling pathway is frequently activated in gastric cancer (GC); however, AKT inhibitors are not effective in unselected GC patients in clinical trials. Mutations in AT-rich interactive domain 1A (ARID1A), which are found in approximately 30% of GC patients, activate PI3K/AKT signaling, suggesting that targeting the ARID1A deficiency-activated PI3K/AKT pathway is a therapeutic candidate for ARID1A-deficient GC.
Methods
The effect of AKT inhibitors was evaluated using cell viability and colony formation assays in ARID1A-deficient and ARID1A knockdown ARID1A-WT GC cells as well as in HER2-positive and HER2-negative GC. The Cancer Genome Atlas cBioPortal and Gene Expression Omnibus microarray databases were accessed to determine the extent of dependence of GC cell growth on the PI3K/AKT signaling pathway.
Results
AKT inhibitors decreased the viability of ARID1A-deficient cells and the inhibitory effect was greater in ARID1A-deficient/HER2-negative GC cells. Bioinformatics data suggested that PI3K/AKT signaling plays a greater role in proliferation and survival in ARID1A-deficient/HER2-negative GC cells than in ARID1A-deficient/HER2-positive cells, supporting the higher therapeutic efficacy of AKT inhibitors.
Conclusions
The effect of AKT inhibitors on cell proliferation and survival is affected by HER2 status, providing a rationale for exploring targeted therapy using AKT inhibitors in ARID1A-deficient/HER2-negative GC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Advanced-stage or recurrent gastric cancer (GC) has a poor prognosis, with high rates of morbidity and mortality worldwide [1, 2]. Despite the development of effective combination treatments for GC, including cytotoxic agents and immune checkpoint inhibitors (ICIs) against programmed cell death 1 (PD-1), the clinical outcome of patients with GC remains poor [3]. Recently developed ICIs have shown increased efficacy, particularly in GC patients with a combined positive score [CPS, which measures the number of programmed cell death ligand 1 (PD-L1) expressing cells] ≥ 5, although not all GC patients benefit from ICIs [4]. Therefore, identifying actionable and druggable targets is necessary to develop effective molecular-targeted therapies.
Large-scale analyses performed by the consortium of The Cancer Genome Atlas (TCGA) and Asian Cancer Research Group led to the classification of GC into molecular subtypes and indicated that GC is a very heterogeneous cancer [5, 6]. The most extensively studied actionable and druggable alteration for targeted therapy in GC is human epidermal growth factor receptor 2 (HER2) overexpression [7]. HER2 overexpression is caused by the amplification of v-erb-bs avian erythroblastic leukemia viral oncogene homolog 2 (ERBB2), which is detected in 15–25% of GC cases [8, 9]. HER2 overexpression promotes cell growth and differentiation in GC by activating the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) and the mitogen-activated protein kinase (MAPK) downstream signaling pathways [10]. Because the addition of trastuzumab to first-line chemotherapy improves overall survival in patients with HER2-positive metastatic GC, HER2 status needs to be determined before the design of first-line therapy [8, 11]. Microsatellite instability (MSI)-high or neurotrophic tyrosine receptor kinase (NTRK) are also therapeutic targets in GC, although these gene alterations occur at low frequency and the benefits of targeting them are thus limited to a few patients [12,13,14,15].
We recently proposed a novel therapeutic strategy for AT-rich interactive domain 1A (ARID1A)-deficient GC using enhancer of zeste homolog 2 (EZH2) inhibitors based on the synthetic lethal approach [16]. ARID1A is a subunit of the Switch/Sucrose Non-fermentable (SWI/SNF) chromatin remodeling complex, and ARID1A mutations are found in approximately 30% of GC cases [17, 18]. ARID1A is a haploinsufficient tumor suppressor gene, and its mutations are mainly truncating mutations, which are not considered druggable targets for conventional chemotherapy [5]. Because ARID1A cooperatively suppresses the PI3K/AKT signaling pathway with EZH2 via PIK3IP1, ARID1A-proficient cells maintain cell proliferation homeostasis [16, 19]. Loss of ARID1A disrupts homeostasis in GC cells, leading to the activation of PI3K/AKT signaling. The PI3K/AKT signaling pathway is also activated by gene amplification of PIK3CA and AKT and loss of PTEN in approximately 30–80% of GC cases [20]. It was reported that the higher expression of phosphorylated (p)-AKT was significantly correlated with poor survival of GC patients [21]. Therefore, it is required to develop anti-AKT therapy for patients with GC and several AKT inhibitors have been tested in clinical trials for GC [21]. However, to date, the efficacy of AKT inhibitors in GC remains low, underscoring the need to identify specific therapeutic or predictive biomarkers for appropriate patient selection [22].
In the present study, we investigated the efficacy of AKT inhibitors against ARID1A-deficient GC cells in vitro to develop an effective targeted therapy. Because HER2 expression plays a role in the efficacy of AKT inhibitors, we evaluated the effect of AKT inhibitors against ARID1A-deficient GC cells stratified by HER2 status using cell experiments and a bioinformatics approach using public databases to elucidate the mechanisms determining drug efficacy.
Materials and methods
Cell culture
The GC cell lines used in this study were obtained from the American Type Culture Collection (Manassas, VA, USA) and RIKEN BioResource Center (Ibaraki, Japan). We used ARID1A-wild-type (ARID1A-WT)/HER2-negative (AGS and NUGC-4 cells), ARID1A-WT/HER2-positive (MKN7 and NCI-N87 cells), and ARID1A-mutated (ARID1A-MUT)/HER2-negative (MKN45 and NUGC-3 cells) cells. Gene mutation data of GC cell lines were obtained from the Catalogue of Somatic Mutations in Cancer (COSMIC) cell line project (http://www.cancer.sanger.a.uk) and revealed that NUGC-3 cells harbor ARID1A mutation [c.5548dup (Insertion)] leading to loss of ARID1A function [p.D1850Gfs*4 (Insertion–Frameshift)]. ARID1A-MUT/HER2-positive cells are not available. The cell lines were authenticated by short tandem repeat analysis in April 2020 (Promega, Madison, WI, USA) [23]. Cells were cultured in RPMI-1640 medium (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) supplemented with 10% fetal bovine serum, 1% penicillin, and 1% streptomycin. All experiments were performed in triplicate. All experiments were carried out in accordance with the approved study plan and relevant guidelines.
Inhibitors
In this study, three kinds of AKT inhibitors, GDC-0068 (ipatasertib, IC50: 5 nM for AKT1, 18 nM for AKT2, and 8 nM for AKT3), AZD5363 (capivasertib, IC50: 3 nM for AKT1, 8 nM for AKT2, and 8 nM for AKT3), and GSK2110183 (afuresertib, IC50: 0.08 nM for AKT1, 2 nM for AKT2, and 2.6 nM for AKT3) were used (Selleck Chemicals, Houston, TX, USA), as previously described [16]. A MAPK inhibitor, HY-15947 (ravoxertinib, IC50: 3.1 nM for ERK1 and 3.1 nM for ERK2, MedChemExpress, Monmouth, NJ, USA), and HER2 inhibitor, CDS022971 (lapatinib, IC50: 10.8 nM for EGFR and 9.2 nM for ERBB2, Sigma–Aldrich) were also used.
Colony formation assay
The effect of drug treatment on cancer cell survival was evaluated using colony formation assays, as previously described [16]. The number of colonies was counted in three random microscopic fields of view (× 40) for each well. Data are presented as the mean ± SD. The experiments were independently performed at least three times.
Cell proliferation assay
Cell proliferation was evaluated using the WST-cell proliferation assay with the Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies, Inc., Kumamoto, Japan) according to the manufacturer’s specifications. Cells were seeded in 96-well plates at a density of 1 × 104 cells/well 1 day after small interfering RNA (siRNA)-mediated knockdown of ARID1A and incubated at 37 °C. Different doses of inhibitors were added when most cells had attached to the well. After further incubation for 48 h, the cells were treated with 10 µL CCK-8 reagent, incubated at 37 °C for 1–4 h, and the absorbance at 450 nm was measured on a microplate reader. The experiments were independently performed at least three times.
Small interfering RNA transfection
The method used for siRNA transfection was described previously [17]. Briefly, knockdown experiments were performed using siRNA oligos for ARID1A (s15784 and s15785; Thermo Fisher Scientific Inc., Waltham, MA, USA), including two target-specific siRNAs and a control siRNA (negative control #1; Thermo Fisher Scientific) according to the manufacturer’s protocol. During the exponential growth phase, cells were transiently transfected with siRNA at a final concentration of 10 nM using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s protocol. After 48 h of incubation, cells were used for the indicated experiments.
Western blotting
Western blot analysis was performed as previously described [16, 17]. The protein blots were incubated with anti-ARID1A antibody (1:1000; catalog no., #12354; Cell Signaling Technology, Danvers, MA, USA), anti-phosphorylated (p)-AKT Ser473 (1:2000; #4060; Cell Signaling Technology), and anti-AKT (1:2000; #4691; Cell Signaling Technology), overnight at 4 °C, followed by incubation with goat anti-rabbit HRP secondary antibody (Santa Cruz Biotechnology, Dallas, Texas, USA) for 1 h at room temperature. The protein levels were quantified using a primary mouse anti-β-actin antibody (dilution, 1:5000; #sc-69789; Santa Cruz Biotechnology) as the internal loading control for 1 h at room temperature. Bound antibodies were detected using the SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific), visualized by autoradiography (ImageQuant™ LAS 4000 IR MultiColor imager; Fujifilm Corporation, Tokyo, Japan), and quantified with Image J software (Version 1.52 s, National Institutes of Health, Bethesda, MD, USA).
Flow cytometry analysis
Cells were seeded in 6-well plates at a density of 5 × 105 cells/well 1 day after siRNA-mediated knockdown of ARID1A and incubated at 37 °C. GSK2110183 (10 µM) was added when most cells had attached to the well. After further incubation for 48 h, treated cells were analyzed using the fluorescein isothiocyanate-Annexin V apoptosis kit (BD Biosciences, San Jose, CA, USA). The results were analyzed with a BD FACSCanto II flow cytometer (BD Biosciences). Flow cytometry data were analyzed using FlowJo 10.7.1 (FlowJo, Ashland, OR, USA, proprietary commercial software, https://www.fowjo.com/) [24].
TCGA database and GEO microarray data analyses
Gene mutation or expression data of GC patients were obtained from TCGA’s cBioPortal database (http://www.cbioportal.org/), including 709 GC patients selected for the analyses from a multi-omics study [5]. For the expression data, RNA-sequencing data normalized by the RSEM method were used for the analyses. Microarray data were downloaded from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo), as previously described [25]. The expression profiles of GC from three independent cohorts deposited as GSE35809, GSE62254, and GSE57303 were used according to the Affymetrix GeneChip Human Genome U133 Plus 2.0 oligonucleotide array [6, 26, 27]. The normalized expression values were obtained from each dataset and were not processed further. If a gene was represented by multiple probe sets, the expression values of multiple probes were averaged. The PI3K/AKT pathway signature represents the level of activation of PI3K/AKT signaling and includes the expression of 105 genes in the REACTOMR_PI3K_AKT_SIGNALING_IN_CANCER in the Gene Set Enrichment Analysis (GSEA) dataset (https://www.gsea-sigdb.org/gsea/index.jsp).
Patient samples
The study included 401 GC surgical specimens from our previous study with clinicopathological data [16, 17]. The study was approved by the ethics committee of Fukushima Medical University. All patients provided written informed consent.
Immunohistochemical staining and evaluation
Immunohistochemical (IHC) staining for ARID1A and PD-L1 was performed in our previous study [17], and HER2 staining was additionally performed in this study. Anti-ARID1A (#12354; D2A8U; 1:500 dilution; Cell Signaling Technology), anti-HER2 (#4290; 1:500; Cell Signaling Technology), and anti-PD-L1 (#13684; E1L3N; 1:400; Cell Signaling Technology) antibodies were used. Evaluation of ARID1A expression was performed as previously described [28]. HER2 expression was evaluated using the ToGA trial scoring system for surgical specimens as follows: 0, no reactivity or membranous reactivity in < 10% of tumor cells; 1 + , faint or barely perceptible membranous reactivity in at least 10% of tumor cells; 2 + , weak to moderate complete, basolateral or lateral membranous reactivity in at least 10% of tumor cells; 3 + , strong complete, basolateral or lateral membranous reactivity in at least 10% of tumor cells [11]. HER2-positive was defined by a score of 3 + , whereas HER2-negative was defined by a score of 0, 1, or 2.
Statistical analysis
The Mann–Whitney U test was used to determine the differences between the two variables. Spearman’s correlation was used to evaluate the correlations between expression levels. All statistical analyses were performed using GraphPad Prism v7.0 (GraphPad Software Inc., San Diego, CA, USA). All P values were two-sided, and P values < 0.05 were considered statistically significant.
Results
ARID1A-MUT GC cells are sensitive to AKT inhibitors
To determine whether ARID1A deficiency affects the sensitivity of GC cells to AKT inhibitors, six GC cell lines were selected from the COSMIC database [four ARID1A-WT and two ARID1A-MUT cell lines] for cell experiments. To examine the role of the PI3K/AKT signaling pathway, we selected ARID1A-WT and HER2-positive or -negative cells because HER2 overexpression activates PI3K/AKT signaling. The expression of ARID1A was confirmed in each GC cell line, as well as the expression of pAKT and AKT, which are indicators of PI3K/AKT signaling activation and the targets of AKT inhibitors [29]. Consistent with the COSMIC database information, ARID1A expression was positive in ARID1A-WT AGS, NUGC4, MKN7, and NCI-N87 cells, whereas it was negative in ARID1A-MUT MKN45 cells (Fig. 1a). Because ARID1A mutations in ARID1A-MUT NUGC-3 cells result in a heterozygous mutant, even though ARID1A was positively expressed, the product resulted in a non-functional protein in NUGC-3 cells [16]. The induction of pAKT was strongest in ARID1A-MUT NUGC-3 cells, followed by ARID1A-WT/HER2-positive MKN7 and NCI-N87 cells (Fig. 1b). ARID1A-WT/HER2-positive MKN7 and NCI-N87 cells showed higher pAKT expression levels than ARID1A-WT/HER2-negative AGS and NUGC-4 cells, confirming the effect of HER2 overexpression on PI3K/AKT signaling activation.

Effect of AKT inhibitors on the viability of ARID1A-wildtype (WT) or -mutated (MUT) and HER2-negative (−) or -positive (+) gastric cancer (GC) cells. a Western blot analysis of ARID1A, pAKT, and AKT expression in whole cell extracts of six GC cell lines. ARID1A expression was positive in four ARID1A-WT cells and negative in ARID1A-MUT MKN45 cells. ARID1A-MUT NUGC-3 cells show positive ARID1A expression although ARID1A mutations lead to the production of a non-functional protein. β-actin was used as a loading control. b Quantitative analysis of pAKT expression relative to AKT was performed using ImageJ software. c ARID1A-WT and ARID1A-MUT cell lines were subjected to cell proliferation assays, and absorbance was measured at 450 nm (OD450nm). Quantification of cell viability results is shown. The concentrations of AKT inhibitors used were as follows: GDC-0068: 0.08, 0.4, 2 10, and 50 μM; AZD5363: 0.08, 0.4, 2 10, and 50 μM; and GSK2110183: 0.08, 0.4, 2 10, and 50 μM. Each value represents the mean ± SD of three independent experiments. d HER2-negative ARID1A-WT and ARID1A-MUT cell lines were subjected to colony formation assays after treatment for 7–14 days with 2 μM GDC-0068, 2 μM AZD5363, and 2 μM GSK2110183. NC negative control. e Quantification of the number of colonies in all fields of each well in (d). NC negative control, GDC GDC-0068, AZD AZD5363, GSK GSK2110183
To determine the sensitivity of ARID1A-deficient GC cells to AKT inhibitors, the viability and survival of ARID1A-WT and ARID1A-MUT cells were measured in the presence of increasing concentrations of AKT inhibitors. Treatment with three AKT inhibitors, GDC-0068, AZD5363, and GSK2110783 [22], significantly decreased the viability of ARID1A-MUT/HER2-negative MKN45 and NUGC-3 cell lines compared with that in ARID1A-WT cells (Fig. 1c). In HER2-negative cells, cell viability was markedly lower in NUGC-3 cells than in MKN45 cells, confirming that the efficacy of AKT inhibitors is associated with pAKT levels induced by ARID1A deficiency. The effect of AKT inhibitors on decreasing cell viability was stronger in ARID1A-WT/HER2-positive MKN7 and NCI-N87 cells than in ARID1A-WT/HER2-negative AGS and NUGC-4 cells, but not as significant as in ARID1A-MUT/HER2-negative MKN45 and NUGC-3 cells. The moderate reduction of cell viability in ARID1A-WT/HER2-positive MKN7 and NCI-N87 cells may be due to HER2 overexpression rather than to ARID1A mutations. In HER2-positive GC cells, the MAPK signaling pathway is activated in addition to the PI3K/AKT signaling pathway; therefore, AKT inhibitors showed lower efficacy in decreasing cell viability in ARID1A-WT/HER2-positive cells than in ARID1A-MUT/HER2-negative cells. These results suggest that HER2 status needs to be considered when evaluating the selectivity of AKT inhibitors according to ARID1A mutations.
To investigate the role of ARID1A deficiency in the sensitivity to AKT inhibitors, cells without HER2 overexpression were subjected to colony formation assays (Fig. 1d). AKT inhibitors significantly suppressed colony growth in ARID1A-MUT/HER2-negative MKN45 and NUGC-3 cells, whereas this effect was not observed in ARID1A-WT/HER2-negative AGS and NUGC4 cells (Fig. 1e). The selective sensitivity of ARID1A-deficient among HER2-negative GC cells was validated by measuring cell survival in colony formation assays. These results indicate that AKT inhibitors show selectivity towards ARID1A-deficient GC cells in the absence of HER2 overexpression.
Selective sensitivity to AKT inhibitors in ARID1A knockdown HER2-negative GC cells
To determine whether HER2 expression affects the efficacy of AKT inhibition, ARID1A was knocked down using two siRNA oligonucleotides (siARID1A #1 and #2) in ARID1A-WT/HER2-negative and ARID1A-WT/HER2-positive GC cells, and cell viability was measured. In cells with ARID1A knockdown, pAKT was markedly upregulated in ARID1A-WT/HER2-negative GC cells but not in AGS cells treated with siARID1A #2 (Fig. 2a, b). pAKT levels did not change significantly in ARID1A-WT/HER2-positive GC cells subjected to ARID1A silencing (Fig. 2a, b). Next, we compared the effect of AKT inhibitors on cell viability between ARID1A knockdown HER2-negative and -positive cells. The three AKT inhibitors decreased the viability of cells with transient ARID1A knockdown compared with that of control cells in HER2-negative NUGC-4 (Fig. 2c) and AGS cells except in those treated with siARID1A#2 (Fig. 2d), indirectly supporting that the efficacy of AKT inhibitors is almost entirely dependent on pAKT levels in HER2-negative GC cells. Similarly, flow cytometry confirmed that apoptosis is increased in NUGC-4 (Fig. 2e) and AGS cells, except for cells treated with siARID1A#2 (Fig. 2f). As shown in Fig. 2a, d, although siARID1A #2 knocked down ARID1A expression, pAKT was not induced in AGS cells. This exceptional result using siARID1A #2 in Fig. 2 was further support that the efficacy of AKT inhibitors in HER2-negative GC cells depends on pAKT levels. Furthermore, AKT inhibitors did not decrease the viability of HER2-positive MKN7 (Fig. 2g) and NCI-N87 (Fig. 2h) cells with transient ARID1A knockdown. These results indicate that ARID1A-deficient/HER2-negative GC cells are selectively sensitive to AKT inhibitors and suggest that AKT inhibitory therapy could be developed as an additional therapeutic strategy for HER2-negative GC.


Effect of AKT inhibitors on the viability of ARID1A-WT/HER2-negative (−) and -positive (+) cells after knockdown of ARID1A. a Western blot analysis of ARID1A, pAKT, and AKT expression in cells treated with siRNAs against ARID1A (siARID1A #1 and #2 and NC, negative control). β-actin was used as a loading control. b Quantification of pAKT expression relative to AKT was performed using ImageJ software. C, d HER2-negative NUGC-4 (c) and AGS (d) cells were subjected to cell proliferation assays after ARID1A knockdown. NC negative control. Absorbance was measured at 450 nm (OD450nm) and cell viability results were quantified. The concentrations of AKT inhibitors used were as follows: GDC-0068: 0.08, 0.4, 2 10, and 50 μM; AZD5363: 0.08, 0.4, 2 10, and 50 μM; and GSK2110183: 0.08, 0.4, 2 10, and 50 μM. Each value represents the mean ± SD of three independent experiments. ARID1A knockdown cells showed a dose-dependent decrease of cell viability and lower cell viability than the negative control. E, f HER2-negative NUGC-4 (e) and AGS (f) cells subjected to ARID1A knockdown were treated with 10 μM GSK2110183 and apoptosis was measured by flow cytometry. g, h ARID1A knockdown and control HER2-positive MKN7 (g) and NCI-N87 (h) cells were treated with AKT inhibitors and cell proliferation was assessed. Absorbance was measured at 450 nm (OD450nm) and the results were quantified. The concentrations of AKT inhibitors used were as follows: GDC-0068: 0.08, 0.4, 2 10, and 50 μM; AZD5363: 0.08, 0.4, 2 10, and 50 μM; and GSK2110183: 0.08, 0.4, 2 10, and 50 μM. Each value represents the mean ± SD of three independent experiments
The role of PI3K/AKT signaling in cell proliferation and survival is more important in HER2-negative than in HER2-positive GC cells
We recently reported that the synthetic lethal approach using EZH2 inhibitors could be a useful therapeutic strategy in PI3K/AKT signaling-dependent ARID1A-deficient GC [16]. AKT inhibitors suppress cell proliferation and survival in ARID1A-deficient/HER2-negative GC cells (Supplementary Fig. S1a). In HER2-positive GC cells, the MAPK pathway is activated in addition to the PI3K/AKT pathway, and inhibition of PI3K/AKT signaling by AKT inhibitors is thus not sufficient to suppress cell proliferation and survival in ARID1A-deficient/HER2-positive GC cells (Supplementary Fig. S1b).
To evaluate these mechanisms in GC, TCGA and GEO microarray databases were used to evaluate PI3K/AKT signaling activation induced by ARID1A deficiency and HER2 overexpression. Analysis of TCGA data indicated that AKT1 expression is higher in ARID1A-MUT GC than in ARID1A-WT GC, although the difference was not statistically significant (P = 0.185) (Fig. 3a). Next, the level of PI3K/AKT signaling activation, which is known as the PI3K/AKT pathway signature, was analyzed using microarray data from three independent cohorts (GSE35809, GSE62254, and GSE57303) (Fig. 3b). A significant inverse correlation between ARID1A mRNA expression and the PI3K/AKT signaling signature was observed in GSE35809, whereas no correlation was observed in the remaining two. Stratification according to HER2 expression showed that AKT expression was significantly lower in HER2-positive GC than in HER2-negative GC (P < 0.0042) (Fig. 3c). The PI3K/AKT signaling signature showed an inverse correlation with ERBB2 mRNA expression in the three microarray cohorts, which was significant in GSE62254 (P < 0.0001) but not in the other two (P = 0.078 and P = 0.284) (Fig. 3d). In ARID1A-WT GC, AKT1 expression was lower in ARID1A-WT/HER2-positive than in ARID1A-WT/HER2-negative GC (P = 0.009) (Fig. 3e). In HER2-negative GC, although AKT1 expression was not induced by ARID1A deficiency (Fig. 3e), the PI3K/AKT pathway signature values were higher in ARID1A-MUT than in ARID1A-WT GC (P = 0.007) (Fig. 3f), supporting the hypothesis in Supplementary Fig. S1 and despite the small number of samples. These results suggest that PI3K/AKT signaling activation is induced by ARID1A deficiency and affects cell proliferation and survival, especially in ARID1A-deficient/HER2-negative GC. By contrast, the dependence of HER2-positve GC on the PI3K/AKT signaling pathway for cancer cell growth is low, which limits the therapeutic efficacy of AKT inhibitors.

Evaluation of the activation and expression of PI3K/AKT signaling genes stratified by ARID1A mutation or HER2 status. a AKT1 protein expression in GC patients with ARID1A-wildtype (WT) (n = 563) and ARID1A truncating mutation (MUT) (n = 146). Data were obtained from The Cancer Genome Atlas (Project ID; TCGA-STAD) (n = 709). b Correlation of ARID1A mRNA expression with the PI3K/AKT pathway signature in GC patients from three cohorts: GSE35809 (n = 70), GSE62254 (n = 300), and GSE57303 (n = 70). A significant inverse correlation between ARID1A expression and the PI3K/AKT pathway signature was observed in GSE35809, which was not significant in GSE62254 and GSE57303. Data were obtained from the GSE microarray database. c AKT1 protein expression in HER2-negative (−) (n = 610) and HER2-positive (+) (n = 99) GC patients. Data were obtained from The Cancer Genome Atlas (Project ID; TCGA-STAD) (n = 709). d Correlation of ERBB2 mRNA expression with the PI3K/AKT pathway signature in GC patients from GSE62254 (n = 300), GSE35809 (n = 70), and GSE57303 (n = 70). A significant inverse correlation between ERBB2 expression and the PI3K/AKT pathway signature was observed in GSE62254 and a marginal correlation was observed in GSE35809 and GSE57303. Data were obtained from the GSE microarray database. e AKT1 protein expression in GC patients with ARID1A-WT/HER2(+) (n = 95), ARID1A-MUT/HER2(+) (n = 4), ARID1A-WT/HER2(−) (n = 468), and ARID1A-MUT/HER2(−) (n = 142). NS not significant. Data were obtained from The Cancer Genome Atlas (Project ID; TCGA-STAD) (n = 709). f The PI3K/AKT pathway signature was compared among GC patients with ARID1A-WT/HER2(+) (n = 6), ARID1A-MUT/HER2(+) (n = 0), ARID1A-WT/HER2(−) (n = 18), and ARID1A-MUT/HER2(−) (n = 10). Data were obtained from The Cancer Genome Atlas (Project ID; TCGA-STAD) (n = 34). g ARID1A knockdown and control HER2-positive MKN7 and NCI-N87 cells were treated with AKT inhibitors, a MAPK inhibitor, AKT with MAPK inhibitors, and a HER2 inhibitor and cell proliferation were assessed. Absorbance was measured at 450 nm (OD450nm) and the results were quantified. The concentrations of inhibitors used were as follows: GDC-0068: 10 μM; AZD5363: 10 μM; GSK2110183: 10 μM; HY-15947: 25 μM for MKN7 and 10 μM for NCI-N87 cells; CDS022971: 25 μM for MKN7 and 10 μM for NCI-N87 cells. Each value represents the mean ± SD of three independent experiments
To further support the hypothesis that the MAPK pathway is additionally activated in HER2-positive GC in Supplementary Fig. S1, we investigated the viability and survival of HER2-positive GC cells were measured in AKT, MAPK, and HER2 inhibitors (Fig. 3g). Treatment with the MAPK inhibitor (HY-15947) decreased the cell viability in HER2-positive MKN7 and NCI-N87 cells more or as much as the AKT inhibitors. The effect of the combination of AKT and MAPK inhibitors on cell viability in HER2-positive cells was stronger than AKT or MAPK inhibitors alone and, notably, as significant as the HER2 inhibitor (CDS022971). These results suggest that the MAPK pathway is simultaneously activated by HER2 overexpression, decreasing the therapeutic efficacy of AKT inhibitors in HER2-positive cells, and indicate again that the effect of AKT inhibitors in HER2-positive cells is independent of ARID1A expression.
Characteristics of ARID1A-deficient/HER2-negative GC patients
To identify patients who may benefit from AKT inhibitors, GC patients were characterized according to ARID1A and HER2 status. Comparison of ERBB2 copy number alterations between ARID1A-WT and ARID1A-MUT GC tumors from TCGA showed that ERBB2 amplification was less frequent in ARID1A-MUT GC (6%) than in ARID1A-WT GC (16%) (Fig. 4a). Consistently, HER2 overexpression was less frequent in ARID1A-MUT than in ARID1A-WT GC (P < 0.001). However, ERBB2 mRNA expression was not positively correlated with ARID1A mRNA expression in a combined microarray cohort (GSE35809, GSE62254, and GSE57303), which was used to improve statistical power (Fig. 4b). Lastly, we performed IHC staining for HER2 in surgically resected GC patients and added the staining results to the clinicopathological data which was previously characterized according to ARID1A expression (Fig. 4c) [16]. In this study, we categorized 401 GC patients by HER2 and ARID1A expression and found that ARID1A-deficient GC tended to correlate with higher TNM stage and depth of invasion and positive lymphatic and venous invasion, especially in HER2-negative GC, consistent with our previous study (Table 1) [17]. As a result, 66 of 77 (85.7%) ARID1A-deficient and 312 of 324 (93.2%) ARID1A-proficient GC cases showed negative HER2 expression, supporting the potential efficacy of AKT inhibitors. These results do not provide a consistent trend in the association between ARID1A and HER2 status, and further studies are needed to understand which patients benefit from AKT inhibitors.

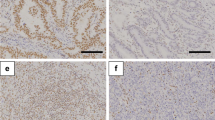
Characteristics of GC patients according to ARID1A and HER2 status. a ERBB2 copy number alterations in GC patients with ARID1A-wildtype (WT) (n = 563) and ARID1A truncating mutation (MUT) (n = 146) (left). HER2 protein expression in GC patients with ARID1A-WT (n = 563) and ARID1A-MUT (n = 146) (right). Data were obtained from The Cancer Genome Atlas (Project ID; TCGA-STAD) (n = 709). b Correlation of ARID1A mRNA expression with ERBB2 mRNA expression in GC patients from a combined cohort including GSE35809 (n = 70), GSE62254 (n = 300), and GSE57303 (n = 70). Data were obtained from the GSE microarray database. c Representative images show immunohistochemical staining of ARID1A-deficient (−), HER2-positive (+), and HER2-negative (−) GC tumors. Scale bar = 250 μm
Discussion
The present study demonstrated that AKT inhibition could be a useful therapeutic strategy for the treatment of ARID1A-deficient GC but limited to HER2-negative GC. This finding may explain the poor efficacy of AKT inhibitors against GC in several clinical trials [22]. Trials evaluating AKT inhibitors alone or in combination with cytotoxic agents showed poor results in unselected GC patients despite the frequent activation of the PI3K/AKT signaling pathway in GC.
Loss of ARID1A activates the PI3K/AKT/mTOR signaling pathway and is thus a potential predictive biomarker of the response to AKT or mTOR inhibitors [30, 31]. In this study, we used three pan-AKT inhibitors to analyze the roles of ARID1A and HER2 in the response of GC cells, thereby providing evidence of the sensitivity of ARID1A-deficient HER2-negative GC to AKT inhibitors, which could be useful to improve biomarker-oriented patient selection. The AKT family includes AKT1, AKT2, and AKT3, which share up to 80% homology at the amino acid level [21]. The pan-AKT inhibitors used in this study, GDC-0068 (ipatasertib), AZD5363 (capivasertib), and GSK2110183 (afuresertib), inhibit all isoforms of AKT. GDC-0068 showed no significant benefit when used in combination with chemotherapy (mFOLFOX6) compared with chemotherapy alone in a randomized phase II trial of first-line therapy for unselected patients with GC [32]. The antitumor effect of GDC-0068 is higher in patients harboring PIK3CA/AKT1/PTEN mutations in several types of cancer; however, this enhanced effect is not observed in patients with GC [33,34,35]. By contrast, AZD5363 shows increased antitumor activity in GC xenograft models harboring either PIK3CA mutations or PTEN loss and in patients with GC harboring PIK3CA mutations [36, 37]. PIK3CA is an important regulator of PI3K/AKT signaling and is often co-regulated with ARID1A, suggesting that PIK3CA mutations enhance the therapeutic effect of AKT inhibitors [38]. Among the GC cells used in this study, ARID1A-WT/HER2-negative AGS and ARID1A-WT/HER2-positive NCI-N87 cells harbor PIK3CA mutations; however, no significant difference in the effect of AKT inhibitors was observed between PIK3CA WT and mutant cell lines.
In recent years, systemic chemotherapy strategies for unresectable advanced or metastatic GC have been designed according to therapeutic target markers such as HER2 [39, 40]. For HER2-positive GC, first-line therapy with trastuzumab plus chemotherapy or subsequent trastuzumab deruxtecan, a novel HER2-targeted antibody–drug conjugate, improves patient overall survival and is becoming the standard treatment regimen [11, 41, 42]. However, systemic chemotherapy remains the first-line treatment for HER2-negative GC, and nivolumab plus chemotherapy was added as standard first-line therapy for tumors expressing PD-L1 CPS ≥ 5 or < 5 under certain conditions, which has extended the median overall survival by approximately 3 months [4, 40]. Targeted therapy is the preferred option as subsequent therapy after first- or second-line therapy, although actionable targets remain to be identified in GC, especially in HER2-negative GC [43].
We concluded that the therapeutic efficacy of AKT inhibitors in ARID1A-deficient/HER2-negative GC is due to the strong dependence of cell proliferation and survival on the activation of the PI3K/AKT signaling caused by ARID1A deficiency. Although PI3K/AKT signaling is also activated by ARID1A deficiency in HER2-positive cells, the dominant pathway activating PI3K/AKT in these cells is constitutive HER2 signaling. In addition, HER2 signaling simultaneously activates the MAPK pathway, which decreases the dependence on the PI3K/AKT pathway and thus the therapeutic efficacy of AKT inhibitors in HER2-positive cells. HER2 inhibitors are therefore the first choice of treatment in HER2-positive GC [40]. In ARID1A-deficient GC with HER2 overexpression, HER2 inhibitors should be used as the first-line therapy. In addition, EZH2 inhibitors based on synthetic lethality are a therapeutic option for ARID1A-deficient/HER2-positive GC as well as ARID1A-deficient/HER2-negative GC [16]. AKT inhibitors or mTOR inhibitors show additive effects in combination with chemotherapy in ARID1A-deficient GC [30, 31]. Because combination therapies are continuously being developed and are expected to have a high therapeutic effect, ARID1A and HER2 status may become a useful predictive biomarker of the response to AKT inhibitors, EZH2 inhibitors, HER2 inhibitors and/or cytotoxic agents especially in HER2-negative GC. One mechanism of trastuzumab resistance is the reactivation of the PI3K/AKT signaling pathway, which suggests that AKT inhibitors could become an additional treatment option for HER2-positive GC as well, although further investigation is required [44].
In this study, ARID1A-deficient/HER2-negative GC cells showed sensitivity to AKT inhibitors, suggesting that AKT inhibition should be reconsidered as a targeted therapy for GC based on ARID1A and HER2 expression status as predictive biomarkers. HER2 is a common marker for the selection of treatment modalities, and the profile of ARID1A mutations can be obtained by comprehensive genomic profiling, which is becoming a standard option in daily clinical practice; further developments are expected in the future.
Data availability
All data generated or analyzed in this study are included in this published article. In silico data in this study are available from public databases.
References
Jim MA, Pinheiro PS, Carreira H, Espey DK, Wiggins CL, Weir HK. Stomach cancer survival in the United States by race and stage (2001–2009): findings from the CONCORD-2 study. Cancer. 2017;123(Suppl 24):4994–5013.
Taniyama Y, Katanoda K, Charvat H, Hori M, Ohno Y, Sasazuki S, et al. Estimation of lifetime cumulative incidence and mortality risk of gastric cancer. Jpn J Clin Oncol. 2017;47:1097–102.
Kono K, Nakajima S, Mimura K. Current status of immune checkpoint inhibitors for gastric cancer. Gastric Cancer. 2020;23:565–78.
Janjigian YY, Shitara K, Moehler M, Garrido M, Salman P, Shen L, et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet. 2021;398:27–40.
Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9.
Cristescu R, Lee J, Nebozhyn M, Kim KM, Ting JC, Wong SS, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med. 2015;21:449–56.
Selim JH, Shaheen S, Sheu WC, Hsueh CT. Targeted and novel therapy in advanced gastric cancer. Exp Hematol Oncol. 2019;8:25.
Dijksterhuis WPM, Verhoeven RHA, Meijer SL, Slingerland M, Haj Mohammad N, de Vos-Geelen J, et al. Increased assessment of HER2 in metastatic gastroesophageal cancer patients: a nationwide population-based cohort study. Gastric Cancer. 2020;23:579–90.
Van Cutsem E, Bang YJ, Feng-Yi F, Xu JM, Lee KW, Jiao SC, et al. HER2 screening data from ToGA: targeting HER2 in gastric and gastroesophageal junction cancer. Gastric Cancer. 2015;18:476–84.
Matsuoka T, Yashiro M. Recent advances in the HER2 targeted therapy of gastric cancer. World J Clin Cases. 2015;3:42–51.
Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–97.
Shinozaki-Ushiku A, Ishikawa S, Komura D, Seto Y, Aburatani H, Ushiku T. The first case of gastric carcinoma with NTRK rearrangement: identification of a novel ATP1B-NTRK1 fusion. Gastric Cancer. 2020;23:944–7.
Akagi K, Oki E, Taniguchi H, Nakatani K, Aoki D, Kuwata T, et al. The real-world data on microsatellite instability status in various unresectable or metastatic solid tumors. Cancer Sci. 2021. https://doi.org/10.1111/cas.14804.
Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13.
Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. 2018;15:731–47.
Yamada L, Saito M, Thar Min AK, Saito K, Ashizawa M, Kase K, et al. Selective sensitivity of EZH2 inhibitors based on synthetic lethality in ARID1A-deficient gastric cancer. Gastric Cancer. 2021;24:60–71.
Ashizawa M, Saito M, Min AKT, Ujiie D, Saito K, Sato T, et al. Prognostic role of ARID1A negative expression in gastric cancer. Sci Rep. 2019;9:6769.
Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43:1219–23.
Saito M, Kono K. Landscape of EBV-positive gastric cancer. Gastric Cancer. 2021;24:983–9.
Sasaki T, Kuniyasu H. Significance of AKT in gastric cancer (review). Int J Oncol. 2014;45:2187–92.
Matsuoka T, Yashiro M. The role of PI3K/Akt/mTOR signaling in gastric carcinoma. Cancers. 2014;6:1441–63.
Kang BW, Chau I. Molecular target: pan-AKT in gastric cancer. ESMO Open. 2020;5: e000728.
Kase K, Saito M, Nakajima S, Takayanagi D, Saito K, Yamada L, et al. ARID1A deficiency in EBV-positive gastric cancer is partially regulated by EBV-encoded miRNAs, but not by DNA promotor hypermethylation. Carcinogenesis. 2021;42:21–30.
Nakajima S, Mimura K, Matsumoto T, Thar Min AK, Ito M, Nakano H, et al. The effects of T-DXd on the expression of HLA class I and chemokines CXCL9/10/11 in HER2-overexpressing gastric cancer cells. Sci Rep. 2021;11:16891.
Nakano H, Saito M, Nakajima S, Saito K, Nakayama Y, Kase K, et al. PD-L1 overexpression in EBV-positive gastric cancer is caused by unique genomic or epigenomic mechanisms. Sci Rep. 2021;11:1982.
Lei Z, Tan IB, Das K, Deng N, Zouridis H, Pattison S, et al. Identification of molecular subtypes of gastric cancer with different responses to PI3-kinase inhibitors and 5-fluorouracil. Gastroenterology. 2013;145:554–65.
Qian Z, Zhu G, Tang L, Wang M, Zhang L, Fu J, et al. Whole genome gene copy number profiling of gastric cancer identifies PAK1 and KRAS gene amplification as therapy targets. Genes Chromosom Cancer. 2014;53:883–94.
George J, Saito M, Tsuta K, Iwakawa R, Shiraishi K, Scheel AH, et al. Genomic amplification of CD274 (PD-L1) in small-cell lung cancer. Clin Cancer Res. 2017;23:1220–6.
Song M, Bode AM, Dong Z, Lee MH. AKT as a therapeutic target for cancer. Cancer Res. 2019;79:1019–31.
Dong X, Song S, Li Y, Fan Y, Wang L, Wang R, et al. Loss of ARID1A activates mTOR signaling and SOX9 in gastric adenocarcinoma-rationale for targeting ARID1A deficiency. Gut. 2022;71:467–78.
Lee D, Yu EJ, Ham IH, Hur H, Kim YS. AKT inhibition is an effective treatment strategy in ARID1A-deficient gastric cancer cells. Onco Targets Ther. 2017;10:4153–9.
Bang YJ, Kang YK, Ng M, Chung HC, Wainberg ZA, Gendreau S, et al. A phase II, randomised study of mFOLFOX6 with or without the Akt inhibitor ipatasertib in patients with locally advanced or metastatic gastric or gastroesophageal junction cancer. Eur J Cancer. 2019;108:17–24.
Sweeney C, Bracarda S, Sternberg CN, Chi KN, Olmos D, Sandhu S, et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): a multicentre, randomised, double-blind, phase 3 trial. Lancet. 2021;398:131–42.
Isakoff SJ, Tabernero J, Molife LR, Soria JC, Cervantes A, Vogelzang NJ, et al. Antitumor activity of ipatasertib combined with chemotherapy: results from a phase Ib study in solid tumors. Ann Oncol. 2020;31:626–33.
Oliveira M, Saura C, Nuciforo P, Calvo I, Andersen J, Passos-Coelho JL, et al. FAIRLANE, a double-blind placebo-controlled randomized phase II trial of neoadjuvant ipatasertib plus paclitaxel for early triple-negative breast cancer. Ann Oncol. 2019;30:1289–97.
Lee J, Kim ST, Kim K, Lee H, Kozarewa I, Mortimer PGS, et al. Tumor genomic profiling guides patients with metastatic gastric cancer to targeted treatment: the VIKTORY umbrella trial. Cancer Discov. 2019;9:1388–405.
Li J, Davies BR, Han S, Zhou M, Bai Y, Zhang J, et al. The AKT inhibitor AZD5363 is selectively active in PI3KCA mutant gastric cancer, and sensitizes a patient-derived gastric cancer xenograft model with PTEN loss to Taxotere. J Transl Med. 2013;11:241.
Wilson MR, Reske JJ, Holladay J, Wilber GE, Rhodes M, Koeman J, et al. ARID1A and PI3-kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. Nat Commun. 2019;10:3554.
Shitara K, Yatabe Y, Matsuo K, Sugano M, Kondo C, Takahari D, et al. Prognosis of patients with advanced gastric cancer by HER2 status and trastuzumab treatment. Gastric Cancer. 2013;16:261–7.
Lordick F, Carneiro F, Cascinu S, Fleitas T, Haustermans K, Piessen G, et al. Gastric cancer: ESMO clinical practice guideline for diagnosis, treatment and follow-up. Ann Oncol. 2022;33:1005–20.
Shitara K, Baba E, Fujitani K, Oki E, Fujii S, Yamaguchi K. Discovery and development of trastuzumab deruxtecan and safety management for patients with HER2-positive gastric cancer. Gastric Cancer. 2021;24:780–9.
Aoki M, Iwasa S, Boku N. Trastuzumab deruxtecan for the treatment of HER2-positive advanced gastric cancer: a clinical perspective. Gastric Cancer. 2021;24:567–76.
Mosele F, Remon J, Mateo J, Westphalen CB, Barlesi F, Lolkema MP, et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: a report from the ESMO Precision Medicine Working Group. Ann Oncol. 2020;31:1491–505.
Diaz-Serrano A, Angulo B, Dominguez C, Pazo-Cid R, Salud A, Jimenez-Fonseca P, et al. Genomic profiling of HER2-positive gastric cancer: PI3K/Akt/mTOR pathway as predictor of outcomes in her2-positive advanced gastric cancer treated with trastuzumab. Oncologist. 2018;23:1092–102.
Acknowledgements
The work was supported by JSPS KAKENHI Grant Number 20K09083 and 21K08675.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no potential conflicts of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sato, T., Saito, M., Nakajima, S. et al. ARID1A deficiency is targetable by AKT inhibitors in HER2-negative gastric cancer. Gastric Cancer 26, 379–392 (2023). https://doi.org/10.1007/s10120-023-01373-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-023-01373-6