
Abstract
Background
In the phase 3 GRID trial, regorafenib improved progression-free survival (PFS) independent of KIT mutations in exons 9 and 11. In this retrospective, exploratory analysis of the GRID trial, we investigated whether a more comprehensive KIT mutation analysis could identify mutations that impact treatment outcome with regorafenib and a regorafenib-induced mutation pattern.
Methods
Archived tumor samples, collected at any time prior to enrollment in GRID, were analyzed by Sanger sequencing (n = 102) and next-generation sequencing (FoundationONE; n = 47). Plasma samples collected at baseline were analyzed by BEAMing (n = 163) and SafeSEQ (n = 96).
Results
In archived tumor samples, 67% (68/102) had a KIT mutation; 61% (62/102) had primary KIT mutations (exons 9 and 11) and 12% (12/102) had secondary mutations (exons 13, 14, 17, and 18). At baseline, 81% of samples (78/96) had KIT mutations by SafeSEQ, including the M541L polymorphism (sole event in 6 patients). Coexisting mutations in other oncogenes were rare, as were mutations in PDGFR, KRAS, and BRAF. Regorafenib showed PFS benefit across all primary and secondary KIT mutational subgroups examined. Available patient-matched samples taken at baseline and end of treatment (n = 41; SafeSEQ), revealed heterogeneous KIT mutational changes with no specific mutation pattern emerging upon regorafenib treatment.
Conclusion
These data support the results of the GRID trial, and suggest that patients may benefit from regorafenib in the presence of KIT mutations and without the selection of particular mutation patterns that confer resistance. The study was not powered to address biomarker-related questions, and the results are exploratory and hypothesis-generating.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The majority of gastrointestinal stromal tumors (GISTs; 70–80%) have mutations in the KIT receptor tyrosine kinase gene resulting in constitutive ligand-independent activation of KIT intracellular signaling [1, 2]. KIT mutations are predominantly found in exon 11 (juxtamembrane domain), with some also found in exon 9 (extracellular domain). Of the remaining GISTs that lack a KIT mutation, a minority (5–10%) have activating mutations in platelet-derived growth factor receptor-α (PDGFRA), while 10–15% of GISTs have no detectable mutations in either KIT or PDGFRA (wild-type GISTs) [2]. Mutations in KIT and PDGFRA are usually mutually exclusive.
Tyrosine kinase inhibitors (TKIs) are the primary therapeutic option for the treatment of metastatic disease [1, 3]. The TKI imatinib demonstrated sustained activity in KIT-mutant GIST, thereby revolutionizing the treatment of GIST [2, 4]. However, many patients develop secondary resistance mutations in KIT, which can arise within the ATP-binding pocket (exons 13 and 14) or activation loop (exons 17 and 18) of the kinase domain [5,6,7]. Following progression on imatinib, patients can go on to receive treatment with the TKI sunitinib, which is active against some imatinib-resistant KIT mutations, but most patients eventually progress.
Regorafenib is an oral TKI that, together with its metabolites, demonstrates activity against a variety of kinases, including wild-type and mutant KIT in vitro and in vivo [8,9,10,11,12]. In the phase 3 GRID trial, regorafenib significantly improved the primary endpoint of progression-free survival (PFS; hazard ratio [HR] 0.27; 95% confidence interval [CI] 0.19–0.39; one-sided P < 0.0001) versus placebo in patients with advanced GIST who previously progressed on imatinib and sunitinib and who were unselected for mutation subtypes [13]. Based on these results, regorafenib was approved as third-line treatment of patients with advanced GIST after imatinib and sunitinib [14, 15], and regorafenib is included as a Category 1 preferred option for these patients in the NCCN Clinical Practice Guidelines in Oncology for GIST [1]. Regorafenib clinical benefit was demonstrated to be independent of detectable primary KIT mutations in exons 9 (HR 0.24; 95% CI 0.07–0.88) and 11 (HR 0.21; 95% CI 0.10–0.46) based on biomarker data at study entry from 66/199 patients in the GRID trial [13].
Using a liquid biopsy approach from specimens collected in the GRID trial, we performed a retrospective exploratory analysis to investigate whether KIT mutations (primary and/or secondary) might impact the treatment benefit of regorafenib, and whether a treatment-induced pattern of mutations could be identified.
Materials and methods
Study design
GRID (NCT01271712) was a randomized, double-blind, placebo-controlled phase 3 trial conducted at 57 centers in 17 countries. Efficacy and safety outcomes of the GRID trial have been reported elsewhere [13]. Briefly, patients with metastatic and/or unresectable GIST who had disease progression with at least prior imatinib and sunitinib were randomized (2:1) to receive oral regorafenib 160 mg once daily or matching placebo, each with best supportive care, for the first 3 weeks of each 4-week cycle. Tumor assessments were performed at baseline, then every 4 weeks for the first 3 months, every 6 weeks for the next 3 months, and every 8 weeks until end of treatment. Patients were treated until disease progression, unacceptable toxicity, or patient decision to withdraw from the trial. In the event of centrally assessed tumor progression, treatment assignment could be unblinded and patients receiving placebo offered to cross over to receive open-label regorafenib. The primary endpoint was PFS (modified Response Evaluation Criteria In Solid Tumors [RECIST] v1.1, by blinded central radiology review) and overall survival (OS) was a secondary endpoint. Biomarker evaluation was an exploratory endpoint.
Biomarker sampling and assays
Archived biopsies and/or plasma samples were collected from patients who provided consent for biomarker analyses. Archived tumor samples were collected at any time prior to the start of GRID, while plasma samples were freshly collected at baseline (i.e., just prior to the start of treatment), and again at the end of treatment (EoT), in the GRID study.
Archived tumor tissue
DNA from archived formalin-fixed, paraffin-embedded tumor tissue specimens was analyzed by Sanger sequencing and next-generation sequencing (NGS). Bi-directional Sanger sequencing was performed on DNA isolated from tumor tissue specimens that were examined by a pathologist and contained at least 50% tumor cells. Tissue specimens judged to be composed of < 50% tumor cells were macro-dissected to enrich for tumor cell content prior to DNA isolation. Tissue specimens that were judged to contain < 50% tumor cells following macro-dissection were not included in the mutational analysis. Exons 9, 11, 13, 14, 17, and 18 in the KIT gene, exons 12, 14, 15, and 18 in the PDGFRA gene, exon 15 in the BRAF gene, and exon 1 in the KRAS gene were sequenced using the Sanger method as previously described [16]. NGS was performed on the FoundationOne panel of 280 tumor genes (Foundation Medicine, Inc.) as previously described [17].
Plasma samples
The amount of human genomic DNA isolated from plasma samples was quantified using a modified version of the human LINE-1 qRT-PCR protocol [18]. DNA is reported in genome equivalents, with one genome equivalent being one haploid human genome weighing 3.3 pg.
Circulating tumor-associated mutations in fresh plasma DNA (ctDNA), collected at baseline, were detected by BEAMing (Beads, Emulsions, Amplification, Magnetics) by Sysmex Inostics GmbH [19]. The BEAMing assays were designed to detect 29 tumor-associated mutations in the KIT gene (exons 9, 11, 17, and 18), 5 in PDGFRA, 1 in BRAF, and 7 in KRAS; the list of mutations analyzed is outlined in Table S1.
Tumor-associated mutations in exons 8–18 in the KIT gene from fresh plasma DNA, collected at baseline and at EoT, were also analyzed by Sysmex Inostics GmbH using a targeted next-generation DNA sequencing approach (SafeSEQ), allowing the detection of de novo missense mutations and insertions/deletions in the region covered by the amplicons. This approach followed the Safe Sequencing protocol as described previously [20], and provides the ability to detect mutant allele frequencies (MAF) down to 0.01% [21] depending on input amount and nucleotide position/change.
Statistical analysis
The biomarker analysis in this study was exploratory and all findings are therefore hypothesis-generating rather than confirmatory. PFS and OS were estimated using the Kaplan–Meier method, with HR and 95% CI calculated using a Cox regression model. No adjustment for multiplicity was performed. Average tumor burden between cfDNA quartiles was compared using a t test.
Results
Patient disposition and demographics
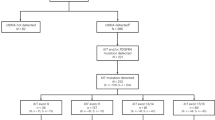
In the GRID trial, 199 patients were randomized to receive regorafenib (n = 133) or placebo (n = 66) between January and August, 2011; one patient in the regorafenib group did not receive treatment [13]. Tissue and plasma samples for biomarker analysis of appropriate quality were available for a subset of patients (Fig. 1). The tissue specimens were from samples that had been collected at various time points prior to study entry, whereas the plasma samples were freshly collected at baseline (i.e., just prior to the start of treatment).

GRID patient subgroups for biomarker analyses. EoT end of treatment
Patient demographics and disease characteristics at baseline in the overall GRID population and BEAMing biomarker cohort were generally similar, while more variation was observed in the SafeSEQ cohort due to the small sample size (Table 1). The PFS and OS treatment benefit in the biomarker subgroups were similar to the overall cohort, apart from slight variation in OS values for the smaller SafeSEQ cohort (Table 2).
Oncogenic mutations in GIST
We analyzed the mutational status in archival tumor tissue and fresh baseline plasma using different mutation-detection methods (Table 3). Initially, archival tumor samples were analyzed by Sanger sequencing for mutations in KIT, PDGFRA, BRAF, and KRAS. A KIT mutation was identified in 67% (68/102) of tumor samples acceptable for analysis (≥ 50% tumor cells); 61% (62/102) had primary mutations, 12% (12/102) had secondary mutations, and 33% (34/102) were KIT wild type. Mutations in the other analyzed oncogenes were rare: PDGFRA mutations were found in 3 samples (3%), activating KRAS mutations were found in 2 samples (2%), one of which did not contain a KIT mutation, and none of the samples had a BRAF mutation. No mutations in KIT, PDGFRA, KRAS, or BRAF were identified in 28% of samples (29/102). The different KIT mutational status in archived biopsies versus plasma samples taken at baseline (Table 3) may partly reflect the differences in prior treatment, with archival biopsies taken prior to treatment with regorafenib and other TKIs (before imatinib/sunitinib treatment) resulting in a lower frequency of secondary KIT mutations.
The archival tumor samples were also analyzed by NGS for the presence of mutations in a number of oncogenes, including KIT. Targeted tumor NGS was performed on 47/102 available samples using the FoundationOne assay on a panel of 280 known oncogenes. A KIT mutation was detected in 85% of samples (40/47) and secondary mutations (in exons 13/14 or 17/18) were present in 26% of samples (12/47). The higher overall mutation rate with NGS, versus Sanger sequencing, may be explained by the higher analytical sensitivity of the NGS method [22]. To confirm this, 30/47 samples were analyzed by both Sanger sequencing and FoundationOne. Discordance between the data sets was found in 17% of samples (5/30), in three of which an additional mutation was detected by NGS. In the 47 samples analyzed by NGS, coexisting oncogenic somatic mutations were rare and identified in genes such as PI3K, MLL2, NF1, NF2, TP53, HRAS, and ErbB4 (1 each; Fig. S1); mutations in PDGFRA were absent from this sample set. Other noteworthy oncogenic driver deletions were identified in CDKN2A (18/47) and RB1 (6/47). Three KIT wild-type samples had mutations in other oncogenes (Fig. S1), but in 4 patients no oncogenic mutations were detectable.
In summary, these results highlight the benefit of using ctDNA liquid biopsies versus archival tumor tissue, and for using sensitive analytical methods.
Baseline circulating DNA levels and association with tumor burden and outcome
To assess if baseline circulating-free plasma DNA (cfDNA) levels could predict tumor burden and outcome, plasma cfDNA levels from 162 patients were analyzed for correlation with tumor burden. Using a quartile analysis, a potential association was identified between high cfDNA levels and increased tumor burden (average tumor burden in the last quartile to that in the first quartile, P = 0.042; Table S2). However, no correlation between baseline cfDNA levels and treatment benefit (PFS or OS) with regorafenib was observed (Table S2).
KIT mutational analysis of baseline circulating DNA
The KIT mutational spectrum in ctDNA was assessed by BEAMing in plasma collected at baseline from 163 of 199 patients (82%). KIT alterations were detected in 58% of patients (94/163); 26% (43/163) were in exons 9 and 11 and were considered to be primary driver mutations, while 47% (77/163) were presumptive secondary resistance mutations (exons 13, 14, 17, and 18) (Table 3). Most of the secondary mutations (64%, 49/77) occurred in exons 17/18 (activation loop), which are associated with exposure and resistance to sunitinib or imatinib [23]. All secondary mutations in patients with a concurrent KIT exon 9 alteration (n = 12) were located in the activation loop. Of note, 40% of patients (31/77) in whom a secondary mutation was identified had multiple secondary mutations, which could either be due to the presence of multiple mutations per lesion or a representation of tumor heterogeneity [24]. Similar to the archival tumor samples, mutations in PDGFRA or BRAF were rare (n = 2 and n = 0, respectively). A KRAS mutation was identified in one of the two patients in whom a KRAS mutation was identified in tumor tissue.
Since the KIT BEAMing assay was designed to detect a predetermined set of mutations, the remaining plasma samples with sufficient material (96 of 163 samples tested by BEAMing) were used to expand coverage of KIT mutations using the targeted NGS SafeSEQ technique. SafeSEQ detects all mutational hotspots and regions harboring presumptive secondary resistance mutations, while also allowing de novo detection of mutations in the respective genes. Using SafeSEQ, a KIT mutation was detected in 81% of samples (78/96) and multiple KIT mutations (up to 13) were found in most samples. Primary mutations were identified in exons 9 (n = 14) and 11 (n = 26), and secondary mutations in exons 13 (n = 15), 14 (n = 7), 17 (n = 43), and 18 (n = 13) (Table 3). As shown in Table S3, an NGS-based approach is particularly useful in regions that are of heterogeneous mutational background, such as deletions in exon 11, or in the presence of a large variety of missense mutations in the same codon (e.g., D820 in exon 17). In several cases, not all mutations detected by BEAMing could be verified by SafeSEQ, which may be due to different levels of input DNA. Presumptive primary mutations in KIT were also detected with SafeSEQ in exon 8 (n = 3), which was not included in the BEAMing analysis. Interestingly, a KIT M541L polymorphism in exon 10 with yet unknown oncogenic function was detected in 24% of patients (23/96), the majority of whom were heterozygous (20/25). In six samples, the M541L variant was the only alteration found. M541L is the most common KIT polymorphism known (rs3822214) with a minor allele frequency of 0.08 in the gnomAD database (https://gnomad.broadinstitute.org/), is classified as benign/likely benign on ClinVar (www.ncbi.nlm.nih.gov/clinvar/), and has been described in the literature as a driver of pediatric mastocytosis [25]. In patients with GIST, the KIT M541L polymorphism has been shown to be associated with a higher risk of metastasis at diagnosis, and with a higher risk of relapse [26, 27]. A higher prevalence of this genotype has also been seen in the disease of higher-risk patients (with tumor duality, un-resectable and/or locally advanced disease, in addition to metastases at diagnosis) compared with patients with localized GIST [27]. In that study, a positive correlation between KIT M541L occurrence and earlier onset of relapse in PDGFRA and wild-type GIST subgroups was found [27].
Correlations between KIT mutational status and patient outcome with regorafenib
Potential association between KIT mutational status and clinical response (PFS) to regorafenib was evaluated. Treatment with regorafenib resulted in longer PFS versus placebo, regardless of whether a secondary mutation was present (HR 0.22, 95% CI 0.12–0.40; P < 0.001) or absent (HR 0.27, 95% CI 0.15–0.49; P < 0.001) in plasma DNA as assessed by BEAMing (Fig. 2A). Regorafenib showed PFS benefit within all mutation subgroups examined across both primary and secondary KIT mutations (Fig. 2B). In addition, regorafenib was associated with PFS benefit versus placebo in the presence (HR 0.046, 95% CI 0.004–0.605) and absence (HR 0.39, 95% CI 0.189–0.805) of the M541L polymorphism in exon 10 of the KIT gene, as determined by SafeSEQ (Fig. S2). Albeit based on a very small data set, our data showed that patients carrying the M541L variant had poor prognosis in the absence of treatment with regorafenib, and had shorter time on treatment with prior imatinib (median 878.5 days) and sunitinib (median 155 days) than those carrying M541 (median 925 and 230.5 days, respectively). This polymorphism was the only alteration found in exon 10.

PFS of regorafenib vs placebo according to A the presence or absence of secondary KIT mutations and B KIT mutation subgroups as determined by BEAMing of plasma DNA (PFS from central assessment). CI confidence interval; HR hazard ratio; INS insertion; PFS progression-free survival
Longitudinal KIT mutational analysis
To evaluate whether treatment with regorafenib results in induction of potential resistance mutations, which is best described by an increase in mutant allele fraction at EoT versus baseline, available individual patient baseline and EoT plasma samples (n = 41) were examined for KIT mutations by SafeSEQ. Duration of regorafenib treatment for patients with matched baseline and EoT samples ranged from 55 to 1131 days (1.8–37.2 months). Although in many cases, there was an increase in the mutant allele fraction across the KIT mutations at EoT versus baseline, the results did not show a trend for an increase of particular mutations that could act as a surrogate for resistance to regorafenib. The mutation analysis revealed a variety of changes in the KIT tumor genotype during treatment that were heterogeneous with no specific mutation pattern or association with outcome (OS) or tumor growth rate (Fig. 3). Some of the changes during treatment include (Table S4): the appearance of an exon 17 secondary mutation (Y823D) in a patient undergoing regorafenib treatment for > 3 years (patient 1); conversion from wild type to exon 17 mutation (D820Y/G) following long-term regorafenib treatment (679 days [22.3 months]; patient 10); disappearance of a primary mutation (exon 11, 557delWK) and strong reduction of secondary mutation allele frequency (D820G) following long-term regorafenib treatment (442 days [14.5 months]; patient 14); strong enrichment of exon 13 mutation (V654A) following long-term regorafenib treatment (433 days [14.2 months]; patient 15); and enrichment of a rare secondary mutation (D820H) following short-term regorafenib treatment (55 days [1.8 months]; patient 27). Interestingly, there was no consistent trend in the conversion of wild type to mutant KIT during treatment, with 7 out of the 11 KIT wild-type patients at baseline remaining wild type at the conclusion of treatment with regorafenib. One of these patients who was treated for 124 days had a PDGFR mutation (A842V). It is noteworthy that, unlike what has been described for imatinib or sunitinib [2, 5, 23, 28], the appearance of secondary mutations was rarely observed (e.g., D820 missense mutation in patient 10). Although intra-patient heterogeneity of biopsy sample-derived mutational profiles cannot be ruled out, these data suggest that progression during regorafenib treatment may be due to other yet-to-be-identified reasons, but unlikely by induction of clones harboring particular KIT exon 17 mutations.

Treatment-induced changes in KIT-MAF and their relationship with OS (A) and TGR (B). Log differences in MAF induced during treatment with regorafenib are plotted on AA position (x-axis) and OS or TGR (y-axis). Negative changes (blue) represent decreasing MAF and positive changes (red) increasing MAF over treatment time. AA amino acid; MAF mutant allele fraction; OS overall survival; TGR tumor growth rate
Discussion
The results presented here confirm the now well-established observation that the majority (up to around 80%) of GISTs harbor KIT mutations, and the rapid translation of these mutational data into effective targeted kinase inhibitor therapies has borne out their importance in GIST pathogenesis [2, 29]. Although being the second-most frequently mutated oncogene in GIST (up to 10%), mutations in PDGFR were rare in this study, which could be due to the fact that PDGFRA- mutated GISTs tend to have a lower risk of recurrence [30]. Preclinical results have shown that regorafenib is active against a number of primary or secondary KIT mutations, and is able to overcome treatment failure after imatinib and sunitinib [8, 10, 11]. The clinical benefit with regorafenib regardless of KIT status observed in the GRID trial [13] supported this hypothesis, and we therefore conducted this retrospective biomarker study to confirm this observation on a molecular level.
Using two analytically highly sensitive liquid biopsy ctDNA assays (BEAMing and SafeSEQ), both of which reliably allow the for the determination of true KIT mutational status in patients with GIST in the third-line setting, the presence of multi-clonal lesions harboring KIT mutations after prior TKI treatment was seen. The analysis did not show any differential efficacy effects for regorafenib on the examined mutational subgroups (primary exon 9 or exon 11 mutations, secondary exon 13/14 or exon 17/18 mutations, or combinations thereof). The activity of regorafenib in the presence of secondary exon 17 KIT mutations has additionally been demonstrated in a small clinical trial [31]. It should be noted that secondary KIT mutations in the activation loop (exons 17/18) were much more prevalent in this study than those in the ATP-binding pocket (exons 13/14). In fact, BEAMing of fresh baseline ctDNA did not identify a single case harboring a secondary mutation in the ATP-binding pocket in the context of a primary KIT exon 9 mutation, whereas secondary mutations in the activation loop were identified in half of these cases. This finding is consistent with a literature report in which cells harboring a primary KIT exon 9 mutation were exposed in vitro to sunitinib, and the resistant clones that grew out were found to harbor secondary KIT mutations located exclusively in the activation loop [23]. In contrast, secondary mutations in the ATP-binding pocket were indeed identified in the context of a primary KIT exon 11 mutation in the current study. Although PFS favored regorafenib (versus placebo) in patients in whom a mutation in the ATP-binding pocket was identified via BEAMing of fresh baseline ctDNA (Fig. 2B), this finding needs to be interpreted with particular caution on account of the relatively small number of these cases comprising this cohort (a correlation of individual secondary mutation with efficacy was not performed in the current analysis due to the low frequency of each individual mutation). SafeSEQ allows the identification of de novo mutations at a detection level (< 0.1% MAF [21]) that is not reached by hybrid capture NGS methods, which typically detect mutations at the 1% level [32]. The approach taken in this study therefore enabled the identification of a large variety of mutations, particularly in exon 17, which provides a sound understanding of the heterogeneity in KIT mutations in this late-line GIST setting. Consequently, this feature makes it particularly attractive to investigate potential treatment-induced changes in KIT mutations. Mutational changes during drug exposure to regorafenib were heterogeneous, with no specific pattern of resistance identified. Both increases and decreases in mutant allele fraction during treatment were observed that were independent of OS or tumor growth rate. The absence of a specific mutation pattern suggests that clonal selection is not a reason as to why these patients progressed on treatment.
Interestingly, this study also showed that a large number of patients were KIT wild type at baseline and remained so during treatment. Furthermore, in the 12 patients where the mutational status at baseline was wild type, the appearance of an activation loop mutation occurred in only one patient, and we therefore propose that the selection of a particular secondary mutation in exon 13/14 or exon 17/18 is not the standard resistance mechanism induced by regorafenib. This result therefore strongly suggests that an outgrowth of particular clones harboring a mutation conferring resistance to regorafenib is not the underlying reason for disease progression. However, it is conceivable that oncogenic alterations in the KIT ectodomain that were not part of this molecular analysis might play a role in driving tumor growth, analogous to what has been observed for the ErbB family of receptor tyrosine kinases [33].
It is important to bear in mind that this was an exploratory and retrospective study, and therefore hypothesis-generating in nature, and the potential impact of low-variant allele frequency mutations is yet to be established. In addition, although highly sensitive, the sequencing techniques used in this analysis have their limitations that have been acknowledged (e.g. in terms of discordance between BEAMing and SafeSEQ). Every effort has been made to mitigate these limitations, such as using more than one technique to analyze samples.
Recently, a variety of novel TKIs have entered clinical development for GIST. One of these is crenolanib, a novel inhibitor of type III receptor tyrosine kinases, which is currently in phase 3 development for PDGFRA D842V mutant GIST (NCT02847429) [34]. Another inhibitor, avapritinib (BLU-285), was developed as a selective inhibitor for mutant KIT and recently received FDA approval for the treatment of advanced GIST harboring a PDGFRA exon 18 mutation [11, 35]. A third inhibitor, ripretinib (DCC-2618), was designed to inhibit the full spectrum of mutant KIT and PDGFRA kinases in cancers [10]. Following encouraging results in a phase 1 trial [36], ripretinib is being evaluated versus sunitinib in a phase 3 trial in GIST following imatinib (NCT03673501). The ctDNA analysis technologies employed in this study may also be useful in the study of potential associations of mutational status with response in these ongoing studies. It remains to be seen if KIT mutation-specific inhibitors offer superiority over unselective inhibitors, such as regorafenib, or whether combinations of agents with complementary activity offers the best solution to combat resistance [12].
In summary, these data suggest that regorafenib may be able to secure long-term treatment benefit by avoiding manifestation of selective resistance mutations and support the positive outcome results of the GRID trial.
Data availability
Availability of the data underlying this publication will be determined later according to Bayer’s commitment to the EFPIA/PhRMA “Principles for responsible clinical trial data sharing”. This pertains to scope, time point and process of data access. As such, Bayer commits to sharing upon request from qualified scientific and medical researchers patient-level clinical trial data, study-level clinical trial data, and protocols from clinical trials in patients for medicines and indications approved in the United States (US) and European Union (EU) as necessary for conducting legitimate research. This applies to data on new medicines and indications that have been approved by the EU and US regulatory agencies on or after January 01, 2014. Interested researchers can use www.clinicalstudydatarequest.com to request access to anonymized patient-level data and supporting documents from clinical studies to conduct further research that can help advance medical science or improve patient care. Information on the Bayer criteria for listing studies and other relevant information is provided in the Study sponsors section of the portal. Data access will be granted to anonymized patient-level data, protocols and clinical study reports after approval by an independent scientific review panel. Bayer is not involved in the decisions made by the independent review panel. Bayer will take all necessary measures to ensure that patient privacy is safeguarded.
References
National Comprehensive Cancer Network Inc. 2020. Clinical Practice Guidelines in Oncology (NCCN Guidelines)—Gastrointestinal Stromal Tumors (GISTs). Version 1. 2021 (2020)
Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11:865–78.
Casali PG, Abecassis N, Bauer S, Biagini R, Bielack S, Bonvalot S, et al. Gastrointestinal stromal tumours: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29:iv68–78.
Demetri GD, Von Mehren M, Blanke CD, Van Den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–80.
Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11:4182–90.
Heinrich MC, Corless CL, Blanke CD, Demettri GD, Joensuu H, Roberts PJ, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24:4764–74.
Wardelmann E, Merkelbach-Bruse S, Pauls K, Thomas N, Schildhaus H, Heinicke T, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006;12:1743–9.
Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schutz G, et al. Regorafenib (BAY 73–4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129:245–55.
Zopf D, Fichtner I, Bhargava A, Steinke W, Thierauch K, Diefenbach K, et al. Pharmacologic activity and pharmacokinetics of metabolites of regorafenib in preclinical models. Cancer Med. 2016;5:3176–85.
Smith BD, Kaufman MD, Lu W-P, Gupta A, Leary CB, Wise SC, et al. Ripretinib (DCC-2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug-resistant KIT and PDGFRA variants. Cancer Cell. 2019;35:738–51.e9.
Evans EK, Gardino AK, Kim JL, Hodous BL, Shutes A, Davis A, et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci Transl Med. 2017;9(414):eaao1690.
Serrano C, Mariño-Enríquez A, Tao DL, Ketzer J, Eilers G, Zhu M, et al. Complementary activity of tyrosine kinase inhibitors against secondary kit mutations in imatinib-resistant gastrointestinal stromal tumours. Br J Cancer. 2019;120:612–20.
Demetri GD, Reichardt P, Kang Y-K, Blay J-Y, Rutkowski P, Gelderblom H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295–302.
Stivarga (regorafenib) Summary of Product Characteristics. Bayer AG (2019). https://www.ema.europa.eu/en/documents/product-information/stivarga-epar-product-information_en.pdf. Accessed June 14, 2021
Stivarga (regorafenib) Prescribing Information. Bayer HealthCare Pharmaceuticals (2020). https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/203085s011lbl.pdf. Accessed June 14, 2021
Tsiatis AC, Norris-Kirby A, Rich RG, Hafez MJ, Gocke CD, Eshleman JR, Murphy KM. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: diagnostic and clinical implications. J Mol Diagn. 2010;12:425–32.
Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–31.
Rago C, Huso DL, Diehl F, Karim B, Liu G, Papadopoulos N, et al. Serial assessment of human tumor burdens in mice by the analysis of circulating DNA. Cancer Res. 2007;67:9364–70.
Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci USA. 2005;102:16368–73.
Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108:9530–5.
Sysmex. SafeSEQ panels and custom solutions. https://cdn2.hubspot.net/hubfs/5871980/SafeSEQ_Technology_fact_sheet_web.pdf. Accessed June 14, 2021.
Arsenic R, Treue D, Lehmann A, Hummel M, Dietel M, Denkert C, Budczies J. Comparison of targeted next-generation sequencing and Sanger sequencing for the detection of PIK3CA mutations in breast cancer. BMC Clin Pathol. 2015;15:20.
Guo T, Hajdu M, Agaram NP, Shinoda H, Veach D, Clarkson BD, et al. Mechanisms of sunitinib resistance in gastrointestinal stromal tumors harboring KITAY502-3ins mutation: an in vitro mutagenesis screen for drug resistance. Clin Cancer Res. 2009;15:6862–70.
Namlos HM, Boye K, Mishkin SJ, Barøy T, Lorenz S, Bjerkehagen B, et al. Noninvasive detection of ctDNA reveals intratumor heterogeneity and is associated with tumor burden in gastrointestinal stromal tumor. Mol Cancer Ther. 2018;17:2473–80.
Foster R, Byrnes E, Meldrum C, Griffith R, Ross G, Upjohn E, et al. Association of paediatric mastocytosis with a polymorphism resulting in an amino acid substitution (M541L) in the transmembrane domain of c-KIT. Br J Dermatol. 2008;159:1160–9.
Brahmi M, Alberti L, Dufresne A, Ray-Coquard I, Cassier P, Meeus P, et al. KIT exon 10 variant (c.1621 A > C) single nucleotide polymorphism as predictor of GIST patient outcome. BMC Cancer. 2015;15:780.
Jasek K, Grendar M, Stanclova A, Malicherova B, Kasubova I, Burjanivova T, et al. Prevalence and significance of M541L single nucleotide polymorphism in the central European cohort of gastrointestinal stromal tumor patients. J Cancer Res Clin Oncol. 2020;147:1203–15.
Desai J, Shankar S, Heinrich MC, Fletcher JA, Fletcher CD, Manola J, et al. Clonal evolution of resistance to imatinib in patients with metastatic gastrointestinal stromal tumors. Clin Cancer Res. 2007;13:5398–405.
Antonescu C. What lessons can be learned from the GIST paradigm that can be applied to other kinase-driven cancers. J Pathol. 2011;223:251–61.
Szucs Z, Thway K, Fisher C, Bulusu R, Constantinidou A, Benson C, et al. Molecular subtypes of gastrointestinal stromal tumors and their prognostic and therapeutic implications. Future Oncol. 2017;13:93–107.
Yeh C-N, Chen M-H, Chen Y-Y, Yang C-Y, Yen C-C, Tzen C-Y, et al. A phase II trial of regorafenib in patients with metastatic and/or a unresectable gastrointestinal stromal tumor harboring secondary mutations of exon 17. Oncotarget. 2017;8:44121–30.
Stetson D, Ahmed A, Xu X, Nuttall BRB, Lubinski TJ, Johnson JH, et al. Orthogonal comparison of four plasma NGS tests with tumor suggests technical factors are a major source of assay discordance. JCO Precis Oncol. 2019. https://doi.org/10.1200/PO.18.00191
O’Connor M, Nicolaides T, Zhang J, Flohr A, Iacone R, Mayweg AV, Epstein DM, Buck E. Abstract LB-111: epidermal growth factor receptor oncogenes expressed in glioblastoma are activated as covalent dimers and exhibit unique pharmacology. Cancer Res. 2019. https://doi.org/10.1158/1538-7445.AM2019-LB-111.
Heinrich MC, Griffith D, McKinley A, Patterson J, Presnell A, Ramachandran A, Debiec-Rychter M. Crenolanib inhibits the drug-resistant PDGFRA D842V mutation associated with imatinib-resistant gastrointestinal stromal tumors. Clin Cancer Res. 2012;18:4375–84.
Ayvakit (avapritinib) Prescribing Information. Blueprint Medicines Corporation (2020). https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212608s000lbl.pdf. Accessed June 14, 2021.
George S, Heinrich MC, Razak ARA, Chi P, Gordon MS, Ganjoo KN, et al. Mutation profile of drug resistant gastrointestinal stromal tumor (GIST) patients (pts) enrolled in the phase 1 study of DCC-2618. J Clin Oncol. 2018;36(15s):11511. https://doi.org/10.1200/JCO.2018.36.15_suppl.11511.
Acknowledgements
We would like to thank the patients, their families, and GRID investigators. We would also like to thank Kaja Wieghardt, Annette Nocon, Vanessa A. Van Rahden, Kristin Stieler, Frank Holtrup, and Frank Diehl (Sysmex Inostics GmbH) for scientific discussion and lab work execution. Editorial assistance in the preparation of his manuscript was provided by OPEN Health Medical Communications (London, UK), with financial support from Bayer.
Funding
This study was funded by Bayer.
Author information
Authors and Affiliations
Contributions
MJ, GDD, IK, and MT conceived and designed the study. All authors contributed to the collection, analysis, and/or interpretation of the data. CK performed the statistical analysis. GB, DHM, and JF performed the mutation analyses. All authors contributed to the drafting and/or critically revising the manuscript, and all authors approved the final version for submission.
Corresponding author
Ethics declarations
Conflict of interest
MJ, CK, IK, and GB are employees of Bayer, and CK owns stock in Bayer.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jeffers, M., Kappeler, C., Kuss, I. et al. Broad spectrum of regorafenib activity on mutant KIT and absence of clonal selection in gastrointestinal stromal tumor (GIST): correlative analysis from the GRID trial. Gastric Cancer 25, 598–608 (2022). https://doi.org/10.1007/s10120-021-01274-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-021-01274-6