
Abstract
Background
Epigenetic aberrations of tumor suppressor genes (TSGs), particularly DNA methylation, are frequently involved in the pathogenesis of gastric cancer (GC). Through a methylome study, we identified eIF4EBP3 as a methylated gene in GC. However, the role of eIF4EBP3 in GC progression has not been explored.
Methods
The expression and promoter region methylation of eIF4EBP3 in GC and healthy tissues were analyzed in public datasets. eIF4EBP3 expression in GC was detected by semi-quantitative RT-PCR, western blot and immunohistochemistry. We also studied epigenetic alterations and functions in GC. The effects of eIF4EBP3 on cell proliferation, migration and invasion were conducted by functional experiments in vitro and in vivo. Label-free proteomic analysis was applied to identify targets of eIF4EBP3.
Results
The expression level of eIF4EBP3 was downregulated in gastric cancer due to promoter region methylation, and was associated with poor survival and tumor progression. Ectopic expression of eIF4EBP3 significantly inhibited tumor cell growth, migration and invasion both in vitro and in vivo. Label-free proteomic analysis indicated eIF4EBP3 downregulated the protein level of β-catenin, which was confirmed by western blot. Overexpression of β-catenin reversed the inhibitory effects of eIF4EBP3 on cell growth and migration, indicating that eIF4EBP3 acts on GC cells by targeting the eIF4E/β-catenin axis.
Conclusion
These results suggest that eIF4EBP3 is a novel TSG methylated in gastric cancer that may play important roles in GC development and liver metastasis and indicate eIF4EBP3 as a potential metastasis and survival biomarker for GC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gastric cancer (GC) is the fifth most frequently diagnosed cancer and the third leading cause of cancer-related death among 36 cancers; over one million new cases of GC were diagnosed in 2018 with an estimated 783,000 deaths worldwide [1]. Although the incidence rates in North America and northern Europe are generally low, the rates are markedly elevated in Eastern Asia, particularly in South Korea, Japan and China [1, 2]. Furthermore, during the last 2 decades, over 40% of GC patients show metastases at initial diagnosis [3]. Liver is the most common site of distant metastasis in GC. Approximately, 4–14% of GC cases have synchronous liver metastases, and the proportion of cases with metachronous liver metastases is up to 37% following radical gastrectomy [4]. Despite various therapeutic measures to treat GC, including surgical resection and chemotherapy regimens, the prognosis of GC patients remains dismal, with an overall 5-year relative survival rate of only 20% in most areas worldwide [5, 6]. Therefore, better understanding of the molecular mechanisms of tumorigenesis and liver metastasis in GC would has significant clinical impact.
Genetic and epigenetic aberrations of tumor-related genes have been thought to play a crucial role in GC development [7, 8]. Among epigenetic alterations, DNA methylation modification has been associated with oncogenesis and cancer progression in GC [9, 10]. Thus, identification of novel tumor suppressor genes (TSGs) targeted by promoter hypermethylation in GC may uncover epigenetic mechanisms for the inactivation of tumor suppressive pathways. These discoveries may also identify potential biomarkers and therapeutic targets for tumor diagnosis, treatment and prognosis in patients with GC.
We searched for candidate TSGs in digestive tract tumors through an epigenomic (CpG methylome) study and massive expression profiling and identified eIF4EBP3 as a new methylated gene in GC (data not shown). eIF4EBP3 is located on chromosome 5q31.3 and is the last identified member of the family of mammalian eukaryotic translation initiation factor 4E binding proteins (eIF4EBPs), which includes three 4E-BP homologues in mammals: 4EBP1, 4EBP2 and 4EBP3 [11, 12]. eIF4EBPs bind to eukaryotic translation initiation factor 4E (eIF4E) and regulate its assembly into EIF4F, the multi-subunit translation initiation factor that recognizes the mRNA cap structure [13,14,15]. All three 4EBPs are thought to negatively regulate eIF4E, but only 4EBP1and 4EBP2 are regulated in a similar way to inhibit cell growth [12, 16, 17]. The details of how eIF4EBP3 binds to eIF4E and is regulated in the cell remain enigmatic, nevertheless, it can be assumed that 4EBP3 may also play an important role in carcinogenesis.
The function of eIF4EBP3 in normal or tumor cells has been rarely reported over the last 2 decades. A recent study demonstrated that the expression of 4EBP3, but not that of 4EBP1 or 4EBP2, was transcriptionally induced during prolonged mTORC1 (mammalian target of rapamycin complex 1) inhibition, revealing 4EBP3 as an important effector of mTORC1 and a predictive biomarker of therapeutic response to prolonged remedy with mTOR-targeting drugs in cancer [18]. Another study identified eIF4EBP3L (the zebrafish homolog of eIF4EBP3) as a key mTOR-dependent regulator of muscle fiber size in response to activity [19]. Additionally, overexpression of eIF4EBP3 has been confirmed to induce apoptosis and attenuate cell proliferation in cervical cancer cells in vitro, indicating its anti-oncogenic potential [20]. These results suggest that eIF4EBP3 may play significant roles in the normal function of cells and the development of tumors. However, the alterations and functions of eIF4EBP3 in gastric cancer have not been studied.
eIF4EBP3 is ubiquitously expressed in human adult tissues, with relatively higher expression in the stomach. Based on our preliminary study showing that eIF4EBP3 was downregulated by hypermethylation in GC cell lines, we explored the inactivation of eIF4EBP3 by promoter methylation and its functions and mechanisms in the development of GC.
Materials and methods
Public cancer database analysis
eIF4EBP3 mRNA expression analysis in gastric cancer tissues
The expression analysis of eIF4EBP3 in gastric cancer specimens was based on data retrieved from oncomine gene expression array database (www.oncomine.org), TCGA database, and GEO database. There is only one mRNA expression database for GC (Cho Gastric) in the oncomine database. The expression data of elF4EBP3 were downloaded for further analysis. Level 3 RNA-sequencing data of GC were downloaded from The Cancer Genome Atlas (TCGA) data portal (https://portal.gdc.cancer.gov/). For the GEO database (http://www.ncbi.nlm.nih.gov/geo/), database was collected in accordance with the following conditions: (1) containing at least 10 cases of normal tissues. (2) Expression data of elF4EBP3 could be successfully extracted. And, four gastric cancer datasets were selected : GSE29272, GSE33651, GSE51575 and GSE55696. Considering the fact that H. pylori has been proved to play critical role in intestinal-type gastric cancer pathogenesis, we searched “gastric cancer and eIF4EBP3 and Helicobacter pylori” in GEO profiles and obtained two additional datasets (GSE10262 and GSE27411) containing eIF4EBP3 expression data in gastric precancerous lesions. Ultimately, eight public datasets were analyzed, with details shown in Table S1. R software (http://www.r-project.org/) was applied to extract and normalize the mRNA expression data from these databases, and then the statistical analyses were performed using GraphPad Prism software version 6.0 (GraphPad Software Inc.).
Transcriptome analysis of eIF4EBP3
To explore the molecular mechanism contributing to the aberrant expression of eIF4EBP3 in gastric cancer, the correlations among mRNA expression of eIF4EBP3 and somatic mutation, copy number variation and DNA methylation were analyzed using online database cBioPortal (www.cbioportal.org) [21], MEXPRESS (https://mexpress.be) [22], and CCLE database (https://portals.broadinstitute.org/ccle/home) [23].
Cell lines, tumor and normal control tissues
The human GC cell lines AGS, BGC823, MKN28, HGC27, SGC7901, MKN45 and KATOIII were purchased from the Cell Bank of Shanghai Institutes for Biological Sciences, the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (Gibco Invitrogen CA, USA), 100 units/mL penicillin, and 100 mg/mL streptomycin at 37 °C, with 5% CO2 in a humidified incubator.
A total of 151 gastric cancer patients who underwent surgery between February 2004 and June 2006 at the Sir Run Run Shaw Hospital (Hangzhou, Zhejiang, China) were enrolled, excluding those who received pre-operative chemotherapy. These patients provided 151 paraffin blocks of GC and matched adjacent normal tissues (ANT) for immunohistochemistry (IHC). This study was approved by the ethics committee of Sir Run Run Shaw Hospital, Zhejiang University.
5-Aza-2’-deoxycytidine (5-Aza) and trichostatin A (TSA) treatment
Cells were treated with 10 μM demethylating agent, 5-Aza (Sigma-Aldrich, St Louis, MO, USA) for 3 days and then further treated with 100 nM histone deacetylase inhibitor trichostatin A (TSA; Cayman Chemical Co., Ann Arbor, MI, USA) for additional 16 h, as described previously [24]. After treatment, cells were harvested for DNA and RNA extraction.
Bisulphite treatment and promoter methylation analysis
Bisulphite modification of DNA, methylation-specific PCR (MSP-PCR) and bisulphite genome sequencing (BGS) were carried out as described previously [24, 25]. MSP-PCR and BGS primers are listed in Table S2.
eIF4EBP3 expressing plasmid and transfection
Gastric cell line MKN45 and SGC7901 were transfected with pCMV6 Entry-eIF4EBP3 plasmid or empty vector (pCMV6 Entry-mock; Origene, MD, USA), using MegaTran 1.0 transfection reagent (Origene), as described previously [26]. Stable eIF4EBP3- and vector-expressing clones were selected for further study.
Cell proliferation assay
Stable transfected cells (1 × 103 for MKN45 and SGC7901) were seeded in triplicate into 96-well plates and allowed to incubate for 24 h. Cell viability was assessed every day according to the Cell Counting Kit-8 (CCK-8) protocol (Dojindo, Kumamoto, Japan).
CFSE proliferation assay
Stable transfected cells were labeled with 5 μM carboxyfluorescein diacetate succinimidyl ester (CFSE) according to the manufacturer’s instruction (biogems, CAT#63310-00).
5-ethynyl-2′-deoxyuridine (EdU) incorporation assay
The rate of cell proliferation was assessed using the Click-iT EdU Imaging Kit (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the manufacturer’s instructions.
Biotin RNA pulldown assay
Cell lysates were prepared by ultrasonication in RIP buffer (150 mM KCl, 25 mM Tris (pH 7.4), 0.5 mM dithiothreitol, 0.5% NP-40, complete protease inhibitors cocktail and RNase inhibitors) and were pre-cleared against streptavidin magnetic beads (Invitrogen). In vitro transcribed biotin-labeled DNA probes (Table S2) were bound to streptavidin magnetic beads, and incubated with cell lysates at 4 °C for 4 h before washing five times with RIP buffer. RNA was isolated using TRIzol.
Animals and tumor model
All in vivo experimental protocols were approved by the animal care committee of Sir Run Run Shaw Hospital, Zhejiang University. Viable Vector/ eIF4EBP3 SGC7901 cells (107 cells in 0.1 ml PBS) were injected into the right groin (six mice/subcutaneous group) or liver (eight mice/intrahepatic group) of 6 week-old female nude mice. At the end of 5th week, tumor weight was measured, and number of metastases was counted. The tumor tissues were fixed prior to IHC staining to assess expressions of eIF4EBP3, β-catenin and Ki67.
Statistical analysis
The statistical analyses were performed using GraphPad Prism software version 6.0 (GraphPad Software Inc.) and SPSS version 20.0 (SPSS Inc.). Differences between groups were determined using the Chi-square test or Student’s t test. Overall survival (OS) after surgery was calculated using the Kaplan–Meier method. Cox stepwise multivariate regression analysis of prognostic factors was performed. A p value of less than 0.05 was considered to be statistically significant.
Results
eIF4EBP3 is downregulated in GC tissues and cell lines by promoter hypermethylation
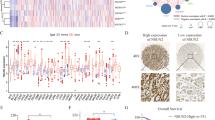
Through analysis of eIF4EBP3 expression in public datasets from gastric cancer patients, we found that eIF4EBP3 was remarkably downregulated in GC tissues and preneoplastic lesions, including low-grade and high-grade intraepithelial neoplasia tissues, compared to normal tissues (Fig. 1a–f). In addition, we observed that eIF4EBP3 mRNA level was also profoundly downregulated in gastric tissues infected with H. pylori or in mouse gastric epithelial progenitor cells treated with H. pylori adenocarcinoma-associated isolates (Fig. 1g, h). This suggests that eIF4EBP3 may play an important role in carcinogenesis of GC.

eIF4EBP3 mRNA expression is downregulated in gastric cancer and precancerous lesions by promoter methylation. a–e Expression of eIF4EBP3 was frequently downregulated in gastric tumor tissues (Tumor) compared with adjacent or normal gastric tissue samples (Normal/Adjacent) in public databases, f–h expression of eIF4EBP3 was downregulated in precancerous lesions of gastric cancer in GEO datasets, i, j relationship between eIF4EBP3 mRNA abundance and copy number alteration and promoter methylation in STAD (cBioPortal), k–l OncoPrint of eIF4EBP3 alterations in STAD. m eIF4EBP3 expression and promoter methylation are negatively correlated, which is confirmed by the Pearson correlation coefficients on the right (MEXPRESS). Data are shown as mean ± SD *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001
To examine the molecular mechanism by which eIF4EBP3 expression is decreased in GC, we used cBioPortal to analyze the types and frequency of eIF4EBP3 mutation in GCs from TCGA and the relationship between its expression and copy number variance and DNA methylation. The results revealed no significant difference between mRNA expression and DNA copy number variation of eIF4EBP3, as shown in Fig. 1i. However, there was a significantly negative correlation between eIF4EBP3 gene expression and DNA methylation (Spearman: − 0.49, p = 5.63e−24) (Fig. 1j). Data in the TCGA database revealed genetic alteration of eIF4EBP3 in 19 of 369 GC patients (5.15%) (Fig. 1k). These alterations included mRNA upregulation in 18 cases (4.88%) and amplification in one case (0.27%); no homozygous deletions and nonsynonymous mutations were detected. The pattern of these transcriptional changes of eIF4EBP3 among various GC subtypes is shown in Fig. 1l.
We next used MEXPRESS to analyze the correlation between DNA methylation of CpG islands in the 5′ promoter region and expression level of eIF4EBP3. The samples shown in Fig. 1m are ordered by expression value. The expression of eIF4EBP3 in GC negatively correlated with the level of DNA methylation in the promoter, which was confirmed by Pearson correlation coefficients (r up to − 0.538, p < 0.001). MEXPRESS analysis also revealed that eIF4EBP3 mRNA expression level was much lower in GC samples than those in control samples (p = 5.736e−05). Thus, these data demonstrate that promoter DNA hypermethylation of eIF4EBP3 may result in downregulation of its mRNA expression, which, to some extent, supports our preliminary study results.
CCLE analysis was consistent with the above results and demonstrated that eIF4EBP3 was present at a relatively lower transcription levels or was absent in GC cell lines (Fig S1a, b). In addition, CpG islands were hypermethylated at multiple sites in these cell lines (Fig S1c).
To validate the expression of eIF4EBP3 and promoter CpG methylation level in GC cell lines, we first examined the protein expression level of eIF4EBP3 by western blot. eIF4EBP3 protein was only detected in KATOIII cells and undetectable in the other six GC cell lines (AGS, BGC823, MKN28, HGC27, SGC7901 and MKN45) (Fig. 2a). RT-PCR showed similar findings with eIF4EBP3 mRNA levels (Fig. 2b). We next evaluated the methylation status of the eIF4EBP3 promoter by MSP-PCR. Full promoter methylation was detected in the six GC lines without detectable eIF4EBP3, but no promoter methylation was observed in KATOIII cells (Fig. 2b). To further confirm that promoter methylation was involved in the downregulation of eIF4EBP3, the seven GC lines were treated with 5-Aza and TSA. Restored expression of eIF4EBP3 was observed in the six GC cell lines with previously undetectable eIF4EBP3, but no changes were observed in KATOIII cells (Fig. 2c). eIF4EBP3 mRNA expression was also decreased in GC samples compared with paired adjacent normal tissues (Fig. 2d). The detailed methylation status of the individual CpG sites within the eIF4EBP3 promoter region was then examined by BGS (Fig. 2e). Consistent with MSP results, full and partial methylation was confirmed in the six GC cell lines, with no methylation detected in KATOIII cells. We further investigated the promoter methylation in 10 pairs of GC specimens. Consistent with the RT-PCR results, BGS revealed full or partial promoter methylation level of eIF4EBP3 in primary GCs, with less methylation in ANT that showed detectable eIF4EBP3 mRNA expression. Together, these results showed that eIF4EBP3 transcription is regulated by promoter CpG methylation.

eIF4EBP3 expression and CpG island methylation in gastric cancer cell lines and primary tumors. a Western blot analysis of eIF4EBP3 in the indicated GC cell lines. β-actin served as loading control, b RT-PCR showed that eIF4EBP3 was expressed in KATOIII cells but absent in the other six GC cell lines tested. DNA methylation was detected in the six GC cell lines, with no DNA methylation in KATOIII cells detected by MSP, c RT-PCR showed that eIF4EBP3 mRNA expression was restored after treatment with demethylation agent 5-aza-2′-deoxycytidine (5-Aza) in GC cells, d transcriptional downregulation of eIF4EBP3 in gastric tumor tissues (Tumor) compared with adjacent normal tissue (ANT) was shown by RT-PCR, e representative BGS results. Cloned BGS-PCR products were sequenced, and each colony is shown as an individual row, representing a single allele of the CpG island. Filled circles represent methylated (different colors indicate coverage) and open circles represent unmethylated CpG sites. M methylated; U unmethylated; MSP methylation-specific PCR; CGI CpG island
Correlation of eIF4EBP3 protein expression with clinicopathological features of GC patients
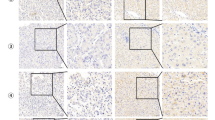
To determine the protein expression of eIF4EBP3 in GC, we next performed IHC analysis on 151 human GC tissues and ANT specimens. Representative results are shown in Fig. 3a, b. eIF4EBP3 was downregulated in primary GC compared with ANT (p < 0.001; Fig. 3c). The correlations between eIF4EBP3 protein expression and the clinicopathological parameters of GC patients are summarized in Table S3. In GC, eIF4EBP3 protein expression was significantly correlated with TNM stage (p = 0.038) and pathologic differentiation (p = 0.009).

Representative immunohistochemical staining of eIF4EBP3 in GC tissues and survival analysis results. a eIF4EBP3 protein was downregulated in tumor tissues and highly expressed mainly in the cytoplasm of paired non-tumor tissues. Corresponding image of hematoxylin and eosin staining section were also shown, b representative immunohistochemical staining of eIF4EBP3 in GC, c eIF4EBP3 was downregulated in primary GC compared with ANT (n= 151, p < 0.001). Kaplan–Meier curves showed that eIF4EBP3 expression is significantly associated with prolonged survival in all GC patients (d) and in patients at an early stage (e). T primary GC tissues; ANT adjacent normal tissue. ***p < 0.001
To examine the impact of eIF4EBP3 overexpression on GC development, we evaluated associations between eIF4EBP3 expression and the clinicopathological features and survival data of GC patients. Kaplan–Meier curves for overall survival (OS) were analyzed. Patients with high eIF4EBP3 expression had significantly longer survival than those with low expression (p < 0.001; Fig. 3d). After stratification by TNM stage, eIF4EBP3 high-expression patients in stage I/II had significantly longer survival than patients with low expression (p = 0.002; Fig. 3e), but no differences were observed among patients in stages III/IV (p > 0.05; data not shown).
Univariate and multivariate Cox regression analyses were performed to identify important prognostic factors of OS (Table S4). eIF4EBP3 expression (p = 0.003) and TNM stage (p < 0.001) were identified as important risk factors for OS. In multivariate Cox analysis, eIF4EBP3 expression (p = 0.022) were found to be independent positive prognostic factors for OS. These findings indicate that eIF4EBP3 predicts good prognosis in patients with GC, especially in the early stages.
Ectopic expression of eIF4EBP3 suppresses GC cell growth in vitro and in vivo
The frequent downregulation of eIF4EBP3 in GC cell lines and tissues suggested that eIF4EBP3 may have a tumor suppressive function and its loss-of-function may promote GC tumorigenesis. To examine this possibility, we generated two GC cell lines (SGC7901 and MKN45) stably overexpressing eIF4EBP3, with empty vector transfection as control. Expression of eIF4EBP3 protein in the stable cell lines was confirmed by western blot analysis (Fig. 4a).

Effects of ectopic eIF4EBP3 expression on the proliferation and invasion of GC cells in vitro and in vivo. a eIF4EBP3 expression in stably transfected cells was confirmed by western blot, b CCK-8 assays examining the effect of ectopic eIF4EBP3 expression on tumor cell growth, c Representative colony formation assay by monolayer culture and quantitative analysis. Values are shown as mean ± SD (d) Representative results of CFSE proliferation assay, e cell proliferation was evaluated using the EdU staining assay, f Ectopic eIF4EBP3 expression in SGC7901 and MKN45 cell lines induced G1/S phase arrest by flow cytometry analysis, g effect of eIF4EBP3 expression on subcutaneous tumor growth in nude mice; quantitative analysis of tumor weight is shown on the right, h transwell assays were performed to evaluate cell migration and invasive abilities in the indicated cell lines, i representative images of wound-healing assay at the indicated times (original magnification: × 100), j Effect of eIF4EBP3 expression on liver metastasis tumor growth in nude mice; quantitative analysis of tumor weight is shown on the right. Data are shown as mean ± SD *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001
Cell proliferation, colony formation and 5-ethynyl-2′-deoxyuridine (EdU) incorporation assays were used to evaluate the effects of ectopic eIF4EBP3 expression on tumor cell growth. CCK-8 results showed that the cell survival rate in eIF4EBP3 stable cell lines was significantly lower than that of control cells (p < 0.001; Fig. 4b). Colony formation assays showed that ectopic expression of eIF4EBP3 inhibited the colony formation ability of GC cells compared with vector-transfected control cells (p < 0.05; Fig. 4c).
CSFE and EdU incorporation assays were used to examine cell proliferation in GC cells. As shown in Fig. 4d, e, the number of proliferating GC cells were decreased in eIF4EBP3 stable cell lines compared with controls. These results indicate that eIF4EBP3 exhibits growth inhibitory activities in GC cells and functions as a tumor suppressor.
To determine the mechanism by which eIF4EBP3 suppresses cell growth, we investigated the effect of eIF4EBP3 on cell cycle and apoptosis by flow cytometry. There were no significant differences in cell apoptosis between eIF4EBP3-transfected SGC7901 cells and controls (data not shown). Flow cytometry revealed cell cycle arrest at the G1-to-S phase transition in both eIF4EBP3-transfected MKN45 and SGC7901 cells (Fig. 4f).
We next evaluated the effects of eIF4EBP3 on the growth of SGC7901 cells in nude mice in vivo. Tumor weight was significantly lower in the eIF4EBP3-transfected nude mouse group compared with the empty vector-transfected group (p < 0.01; Fig. 4g). These results suggest that eIF4EBP3 acts as a tumor suppressor in GC carcinogenesis in vivo.
Ectopic eIF4EBP3 suppresses migration and invasion of GC cells in vitro and liver metastasis in vivo
To evaluate the effects of eIF4EBP3 expression on the migration and invasion of GC cells, transwell and wound-healing assays were performed. Ectopic expression of eIF4EBP3 markedly suppressed cell migration ability in both MKN45 and SGC7901 cells (p < 0.001; Fig. 4h). Matrigel invasion assay also showed that eIF4EBP3 expression significantly impaired the invasiveness of both cell lines (p < 0.05; Fig. 4h). Wound-healing assay was further applied to assess the effect of eIF4EBP3 on cell migration. The results demonstrated that ectopic eIF4EBP3 inhibited migration ability of GC cells (Fig. 4i).
We next evaluated the effects of eIF4EBP3 on the invasive ability of SGC7901 cells in vivo by examining liver metastases in nude mice. Tumor weight was significantly lower in the eIF4EBP3-transfected nude mouse group relative to the empty vector-transfected group (p < 0.01, (Fig. 4j). Notably, intrahepatic metastases were detected in the empty vector-transfected group (3/8, 37.5%), while no intrahepatic metastases were found in the eIF4EBP3-expressing group. These results suggested that eIF4EBP3 acts as a tumor suppressor in GC cells in vivo through suppressing cell proliferation and migration.
Ectopic eIF4EBP3 expression inhibits GC cell growth and migration via the eIF4E/β-catenin axis
To identify the signal transduction pathways that are involved in eIF4EBP3-mediated inhibition of proliferation and invasion of GC cells, we performed quantitative proteomic profiling of SGC7901 cells. Three biological replicates on viable Vector/eIF4EBP3 SGC7901 cells were tested and protein abundance was quantified. A total of 661 proteins were dysregulated, including 304 upregulated proteins (eIF4EBP3/Vector, ratios > 2.0, p < 0.05) and 357 downregulated proteins (ratios < 0.5, p < 0.05) (Fig S2a, b). The top 40 most differentially expressed proteins are listed in Fig. 5a. β-catenin ranked 14th among proteins with the most significant decreased expression. We thus speculated that β-catenin may play an important role in eIF4EBP3-induced inhibition of proliferation and invasion in GC cells.

Ectopic eIF4EBP3 inhibits GC cell growth and migration via the eIF4E/ β-catenin axis, a Label-free proteome analysis showed that β-catenin was significantly downregulated in eIF4EBP3 stably transfected SGC7901 cells compared with empty vector-transfected cells, b correlation between mRNA expressions of eIF4EBP3 and β-catenin in GC tissues in TCGA, c correlation between mRNA expressions of eIF4EBP3 and β-catenin in GC cell pairs, d agarose gel electrophoresis and qPCR showed fewer ribosomes on CTNNB1 mRNA in eIF4EBP3-transfected SGC7901 cells compared with the control group, e representative immunohistochemistry images of eIF4EBP3, β-catenin and Ki67 in tumor tissues of liver metastasis (original magnification: × 200), f Western blot of eIF4EBP3 and β-catenin in tumor tissues of liver metastasis, g Western blot of β-catenin, E-cadherin, slug, and Vimentin in stably transfected GC cell lines, h Western blot of p-eIF4E, eIF4E, GSK3β and p-GSK3β in stably transfected and empty vector-transfected cells lines, i Western blot shows that cyclin D1 expression was downregulated in both stably transfected cell lines, while CDK2, CDK4, and CDK6 were upregulated
We found that β-catenin expression was significantly increased in GC tissues compared with control tissues in the TCGA RNA-seq database (online website GEPIA: http://gepia.cancer-pku.cn/) (Fig S2c). Enrichment analysis of KEGG and GO with β-catenin expression was performed (Fig S2d–f). The most significant pathway involved Adherens junction, gastric cancer and Wnt signaling pathways. We also detected a significant negative correlation between the expression of β-catenin and eIF4EBP3 in TCGA (r2 = − 0.18, p = 0.00023; Fig. 5b), however, no significant correlation was found between the mRNA levels of β-catenin and eIF4EBP3 (Fig. 5c). We used RNA pulldown–qPCR assays to evaluate the ribosome content on β-catenin mRNA and found that fewer ribosomes bound to β-catenin mRNA in the eIF4EBP3-transfected SGC7901 cells than in the control group (Fig. 5d). Western blot confirmed that the expression of β-catenin was decreased in both eIF4EBP3-transfected MKN45 and SGC7901 cells (Fig. 5g). Decreased expression of β-catenin was also confirmed by IHC and western blot assays of liver metastases in nude mice (Fig. 5e, f).
Previous studies have established β-catenin as a crucial molecule in EMT and the Wnt/β-catenin pathway, and its expression is frequently detected in GC. To further explore the relationship between eIF4EBP3 and β-catenin, we investigated the protein levels of EMT markers and Wnt pathway proteins (GSK3β, p-GSK3β, p-EIF4E and eEIF4E). Western blot showed that ectopic eIF4EBP3 expression resulted in decreased β-catenin and slug levels and increased E-cadherin and Vimentin in both eIF4EBP3 stable cell lines compared with empty vector-transfected cells (Fig. 5g). p-GSK3β and p-EIF4E were decreased after ectopic expression of eIF4EBP3 in GC cells (Fig. 5h).
To further elucidate the mechanism of G1/S phase arrest induced by eIF4EBP3, we examined the expression changes of proteins involved in the G1/S phase transition. Western blot showed that the expression of cyclin D1 was downregulated in both eIF4EBP3 stable cell lines compared with controls, while CDK2, CDK4 and CDK6 were upregulated (Fig. 5i). These results suggested that eIF4EBP3 suppresses GC via inhibiting eIF4E-mediated β-catenin translation, which affects the molecular signal changes mediated by β-catenin, including cyclin D1.
Restoring β-catenin protein reverses eIF4EBP3-induced inhibition in GC cells
To further determine the role of β-catenin in eIF4EBP3-mediated inhibition of GC growth and invasion, we overexpressed β-catenin in SGC7901 cells stably expressing eIF4EBP3. Expression of β-catenin was confirmed by western blot (Fig. 6a). We found that the proliferative ability of cells with restored β-catenin expression was greater than controls by CCK-8 assay (p < 0.001; Fig. 6b) and EdU incorporation assay (Fig. 6c). Colony formation assay showed that overexpression of β-catenin significantly improved colony formation ability of eIF4EBP3 stable cell lines overexpressing β-catenin compared with vector-transfected cells (p < 0.001; Fig. 6d). Transwell and wound-healing assays were performed to further evaluate the effects of β-catenin overexpression on the migration and invasion of GC cells. Overexpression of β-catenin increased cell migration and invasion ability in SGC7901 cells (p < 0.01; Fig. 6e). Wound-healing assay further demonstrated that β-catenin inhibited migration ability in eIF4EBP3 stable cell lines (Fig. 6f).

Restoring β-catenin protein reverses eIF4EBP3-induced inhibition in GC cells. a Western blot of β-catenin in stably eIF4EBP3 transfected SGC7901 cells. b, c overexpression of β-catenin protein restored cell proliferation ability in stably eIF4EBP3 expressing cells as shown by CCK-8 and EdU staining assays. d colony formation assay showed that β-catenin promotes colony formation and proliferation in stably eIF4EBP3 expressing cells. e, f expression of β-catenin restores cell migration and invasion ability in stably eIF4EBP3 expressing cells as shown by transwell and wound-healing assays (2 × 105 cells)
Discussion
GC is one of the most common cancers worldwide, with a poor prognosis and limited therapeutic measures [27]. Elevated mRNA translation is often observed in cancers and required for tumor development and progression [28]. In eukaryotes, translation initiation is the rate-limiting step and mediated by the eIF4F complex, which is composed of eIF4E (cap-binding subunit), eIF4A (RNA helicase) and eIF4G (scaffolding protein) [29]. The activity of the eIF4F complex is widely regulated by multiple signaling pathways; however, most of these pathways converge in fine to the target for cap-binding subunit eIF4E [30]. eIF4E is responsible for recognizing and binding the 5′ cap structure of eukaryotic mRNA, and the recognization varies dramatically among mRNAs. eIF4E preferentially binds mRNAs encoding proliferation- and survival-promoting genes such as cyclin D1 and D3, c-myc, VEGF, survivin and Bcl2 genes. Most of these genes have been shown to be important for tumor development and progression [31]. Furthermore, eIF4E has been consistently demonstrated as an oncogene in many types of cancer, including GC [32, 33]. The activity of eIF4E is closely regulated, in part by eIF4EBPs, which sequester eIF4E from the eIF4F complex. Three eIF4EBPs have been identified: eIF4EBP1, eIF4EBP2 and eIF4EBP3 [12]. Based on the demonstrated oncogenic function of eIF4E, eIF4EBPs may play tumor suppressive role in cancer, and this was proven in a previous investigation [17]. eIF4EBP1 and eIF4EBP2 are well studied, while less is known regarding eIF4EBP3 [20]. Moreover, the role of eIF4EBP3 in GC had not yet been reported. Here, we investigated the correlation between eIF4EBP3 level and clinical outcome of GC patients and dissected the function and underlying mechanism of eIF4EBP3 in GC.
Using a public database of GC patients, eIF4EBP3 was found to be downregulated in tumors and preneoplastic lesions. Moreover, GC patients with high level of eIF4EBP3 showed a longer OS than those with low level of eIF4EBP3 (p < 0.05). After stratification by TNM stages, eIF4EBP3 was also a reliable prognosis factor for GC patients in stage I/II. These results suggested that eIF4EBP3 could be an independent positive prognostic factor for GC patients, especially patients in the early stages. Downregulation of eIF4EBP3 in even pre-neoplasms also implies that inhibiting eIF4EBP3 is a prerequisite for GC initiation. The decline of eIF4EBP3 expression was not caused by gene mutation, since no homozygous deletion or nonsynonymous mutation was detected in GC in the cBioPortal and TCGA database. However, hypermethylation at the 5′ promoter region in GC was found by epigenomic screening. Furthermore, eIF4EBP3 expression showed a significantly negative correlation with DNA methylation of CpG islands in the 5′ promoter region. The critical role of epigenetic dysregulation on cancers has been demonstrated in recent decades, and aberrant DNA methylation is the most extensively investigated mechanism in GC [7, 34]. To ascertain if downregulation of eIF4EBP3 in GC was caused by DNA hypermethylation, we assessed the expression of eIF4EBP3 and DNA methylation status in GC cell lines. The expression of eIF4EBP3 was diminished in most GC cell lines, and hypermethylated CpGs were detected in the promoter region of eIF4EBP3. Moreover, suppressing DNA methylation by 5-Aza and TSA can restore the expression of eIF4EBP3 in GC cell lines. Comparable results were also observed in specimens of GC patients. DNA hypermethylation of CpGs islands in GC is detectable in TSGs, such as p16, RUNX3, CDH1 and APC genes [35]. Our results suggested that eIF4EBP3 is another TSG that is suppressed by DNA hypermethylation. Distinct from eIF4EBP3, while the silencing of eIF4EBP1 and eIF4EBP2 in cancer has been mainly attributed to oncogenic signaling pathways [17]. This reflects diverse routes by which tumor cells overcome eIF4EBPs function. These data also imply that reactivating eIF4EBP3 expression by removing hypermethylation in its promoter region could be a potential therapeutic approach for GC treatment.
We also examined the function and underlying mechanism of eIF4EBP3 in GC cell lines. Overexpressing eIF4EBP3 significantly suppressed proliferation and tumor growth of GC cells in vitro and in vivo. MS was performed to clarify downstream effectors of eIF4EBP3. Among the significantly varied signaling pathways induced by eIF4EBP3, we focused on Wnt/β-catenin pathway, a well-known oncogenic pathway in many types of cancers [36]. Overexpressing eIF4EBP3 decreased the protein level of β-catenin, and the tumor suppressive function of eIF4EBP3 was reversed by β-catenin expression. An inverse correlation between eIF4EBP3 and β-catenin was also detected in tumors of GC patients. A previous study reported that eIF4E activates the Wnt/β-catenin pathway by enhancing the translation of β-catenin [37]. A similar mechanism was also detected in our research, with reduced phosphorylated eIF4E and repressed β-catenin translation upon overexpressing eIF4EBP3 in GC cells. Together this suggests that eIF4EBP3 suppresses GC via inhibiting eIF4E-mediated β-catenin translation (Fig. 7).

Model for the mechanism of eIF4EBP3 in the development of GC
Tumor metastasis is a critical obstacle for cancer intervention, and previous studies showed that the Wnt/β-catenin pathway promotes the metastasis of many cancers by EMT [38]. As our results showed that eIF4EBP3 downregulates β-catenin, we evaluated whether eIF4EBP3 could suppress GC metastasis. In vitro studies showed that eIF4EBP3 inhibits the migration and invasion of GC cell lines. EMT-related proteins were also altered by eIF4EBP3 overexpression, including slug, a target of the Wnt/β-catenin pathway [39]. Clinical data have shown that the most common site for GC distant metastasis is the liver, and patients with liver metastasis have a short survival time [40]. Therefore, we investigated the effect of eIF4EBP3 overexpression on liver metastasis in nude mice and the results suggested that eIF4EBP3 inhibits metastasis of GC cells to liver.
Previous studies have elucidated the contribution of eIF4E and eIF4EBP1 in GC [41, 42]. However, the role of eIF4EBP3 in GC has not been reported yet. In the present work, eIF4EBP3 was proven to be a tumor suppressor in GC development; eIF4EBP inhibited the growth of GC cells and its expression was shown to be an independent prognostic factor for GC patients. Moreover, eIF4EBP3 suppressed the metastasis of GC cells to liver. Mechanistically, eIF4EBP3 exhibited tumor suppressive functions through inhibiting eIF4E-mediated β-catenin translation. Silencing of eIF4EBP3 in GC was caused by DNA hypermethylation at its promoter region. However, the factors regulating this process are still unclear and require future studies.
Taken together, we demonstrated that eIF4EBP3, which is frequently downregulated or silenced in GC by DNA hypermethylation, is a tumor suppressor with prognostic and therapeutic value in GC.
Conclusions
In present study, we first elucidated that eIF4EBP3 plays a TSG role in GC, and its downregulation in GC patients was caused by DNA hypermethylation at its 5’-promoter region. eIF4EBP3 inhibits the proliferation, migration and metastasis to liver of GC cells, partly by destabilizing the stability of β-catenin mRNA. Our work sheds lights on targeting eIF4EBP3 for GC prognosis and treatment.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86.
Thomassen I, van Gestel YR, van Ramshorst B, Luyer MD, Bosscha K, Nienhuijs SW, et al. Peritoneal carcinomatosis of gastric origin: a population-based study on incidence, survival and risk factors. Int J Cancer. 2014;134(3):622–8.
D’Angelica M, Gonen M, Brennan MF, Turnbull AD, Bains M, Karpeh MS. Patterns of initial recurrence in completely resected gastric adenocarcinoma. Ann Surg. 2004;240(5):808–16.
Nashimoto A, Akazawa K, Isobe Y, Miyashiro I, Katai H, Kodera Y, et al. Gastric cancer treated in 2002 in Japan: 2009 annual report of the JGCA nationwide registry. Gastric Cancer. 2013;16(1):1–27.
Sun F, Sun H, Mo X, Tang J, Liao Y, Wang S, et al. Increased survival rates in gastric cancer, with a narrowing gender gap and widening socioeconomic status gap: a period analysis from 1984 to 2013. J Gastroenterol Hepatol. 2018;33(4):837–46.
Choi IS, Wu TT. Epigenetic alterations in gastric carcinogenesis. Cell Res. 2005;15(4):247–54.
Padmanabhan N, Ushijima T, Tan P. How to stomach an epigenetic insult: the gastric cancer epigenome. Nat Rev Gastroenterol Hepatol. 2017;14(8):467–78.
Zouridis H, Deng N, Ivanova T, Zhu Y, Wong B, Huang D, et al. Methylation subtypes and large-scale epigenetic alterations in gastric cancer. Sci Transl Med. 2012;4(156):156ra40.
Zeng XQ, Wang J, Chen SY. Methylation modification in gastric cancer and approaches to targeted epigenetic therapy (Review). Int J Oncol. 2017;50(6):1921–33.
Poulin F, Gingras AC, Olsen H, Chevalier S, Sonenberg N. 4E-BP3, a new member of the eukaryotic initiation factor 4E-binding protein family. J Biol Chem. 1998;273(22):14002–7.
Peter D, Igreja C, Weber R, Wohlbold L, Weiler C, Ebertsch L, et al. Molecular architecture of 4E-BP translational inhibitors bound to eIF4E. Mol Cell. 2015;57(6):1074–87.
Alain T, Morita M, Fonseca BD, Yanagiya A, Siddiqui N, Bhat M, et al. eIF4E/4E-BP ratio predicts the efficacy of mTOR targeted therapies. Cancer Res. 2012;72(24):6468–76.
Paku KS, Umenaga Y, Usui T, Fukuyo A, Mizuno A, In Y, et al. A conserved motif within the flexible C-terminus of the translational regulator 4E-BP is required for tight binding to the mRNA cap-binding protein eIF4E. Biochem J. 2012;441(1):237–45.
Siddiqui N, Sonenberg N. Signalling to eIF4E in cancer. Biochem Soc Trans. 2015;43(5):763–72.
Rousseau D, Gingras AC, Pause A, Sonenberg N. The eIF4E-binding proteins 1 and 2 are negative regulators of cell growth. Oncogene. 1996;13(11):2415–20.
Martineau Y, Azar R, Bousquet C, Pyronnet S. Anti-oncogenic potential of the eIF4E-binding proteins. Oncogene. 2013;32(6):671–7.
Tsukumo Y, Alain T, Fonseca BD, Nadon R, Sonenberg N. Translation control during prolonged mTORC1 inhibition mediated by 4E-BP3. Nat Commun. 2016;20(7):11776.
Yogev O, Williams VC, Hinits Y, Hughes SM. eIF4EBP3L acts as a gatekeeper of TORC1 in activity-dependent muscle growth by specifically regulating Mef2ca translational initiation. PLoS Biol. 2013;11(10):e1001679.
Lv KT, Liu Z, Feng J, Zhao W, Hao T, Ding WY, et al. MiR-22-3p regulates cell proliferation and inhibits cell apoptosis through targeting the eIF4EBP3 gene in human cervical squamous carcinoma cells. Int J Med Sci. 2018;15(2):142–52.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):1.
Koch A, De Meyer T, Jeschke J, Van Criekinge W. MEXPRESS: visualizing expression, DNA methylation and clinical TCGA data. BMC Genom. 2015;26(16):636.
Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7.
Ying J, Li H, Seng TJ, Langford C, Srivastava G, Tsao SW, et al. Functional epigenetics identifies a protocadherin PCDH10 as a candidate tumor suppressor for nasopharyngeal, esophageal and multiple other carcinomas with frequent methylation. Oncogene. 2006;25(7):1070–80.
Tao Q, Huang H, Geiman TM, Lim CY, Fu L, Qiu GH, et al. Defective de novo methylation of viral and cellular DNA sequences in ICF syndrome cells. Hum Mol Genet. 2002;11(18):2091–102.
Liu X, Hu X, Kuang Y, Yan P, Li L, Li C, et al. BCLB, methylated in hepatocellular carcinoma, is a starvation stress sensor that induces apoptosis and autophagy through the AMPK-mTOR signaling cascade. Cancer Lett. 2017;1(395):63–71.
Karimi P, Islami F, Anandasabapathy S, Freedman ND, Kamangar F. Gastric cancer: descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol Biomarkers Prev. 2014;23(5):700–13.
Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I. Targeting the translation machinery in cancer. Nat Rev Drug Discov. 2015;14(4):261–78.
Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010;11(2):113–27.
Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136(4):731–45.
Karaki S, Andrieu C, Ziouziou H, Rocchi P. The eukaryotic translation initiation factor 4E (eIF4E) as a therapeutic target for cancer. Adv Protein Chem Struct Biol. 2015;101:1–26.
Graff JR, Konicek BW, Carter JH, Marcusson EG. Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer Res. 2008;68(3):631–4.
Liang S, Guo R, Zhang Z, Liu D, Xu H, Xu Z, et al. Upregulation of the eIF4E signaling pathway contributes to the progression of gastric cancer, and targeting eIF4E by perifosine inhibits cell growth. Oncol Rep. 2013;29(6):2422–30.
Kang GH, Lee S, Kim JS, Jung HY. Profile of aberrant CpG island methylation along the multistep pathway of gastric carcinogenesis. Lab Investig J Tech Methods Pathol. 2003;83(5):635–41.
Li Y, Liang J, Hou P. Hypermethylation in gastric cancer. Clin Chim Acta Int J Clin Chem. 2015;448:124–32.
Krishnamurthy N, Kurzrock R. Targeting the Wnt/beta-catenin pathway in cancer: update on effectors and inhibitors. Cancer Treat Rev. 2018;62:50–60.
Karni R, Gus Y, Dor Y, Meyuhas O, Levitzki A. Active Src elevates the expression of beta-catenin by enhancement of cap-dependent translation. Mol Cell Biol. 2005;25(12):5031–9.
Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36(11):1461–73.
Wu ZQ, Li XY, Hu CY, Ford M, Kleer CG, Weiss SJ. Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc Natl Acad Sci USA. 2012;109(41):16654–9.
Shitara K, Ikeda J, Kondo C, Takahari D, Ura T, Muro K, et al. Reporting patient characteristics and stratification factors in randomized trials of systemic chemotherapy for advanced gastric cancer. Gastric Cancer Off J Int Gastric Cancer Assoc Jpn Gastric Cancer Assoc. 2012;15(2):137–43.
Chen CN, Hsieh FJ, Cheng YM, Lee PH, Chang KJ. Expression of eukaryotic initiation factor 4E in gastric adenocarcinoma and its association with clinical outcome. J Surg Oncol. 2004;86(1):22–7.
Nishi T, Iwasaki K, Ohashi N, Tanaka C, Kobayashi D, Nakayama G, et al. Phosphorylation of 4E-BP1 predicts sensitivity to everolimus in gastric cancer cells. Cancer Lett. 2013;331(2):220–9.
Acknowledgements
We thank Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Funding
This work was supported by the Grants from National Natural Science Foundation of China (81772546, 81902367) and Zhejiang Provincial Natural Science Foundation of China (LQ19H160026, LY19H160057) and Major Scientific and Technological Project of Zhejiang Province (2015C03026).
Author information
Authors and Affiliations
Contributions
XJC, XTH and YFW conceived and designed the study. STZ, SL, ZJL, JJX, TJ, KC, KW, HL, HNY, WQF, JW and GXF performed the experiments. STZ, SL and ZJL performed qPCR, cell culture, H&E and IHC. STZ, SL, KC, KW, and ZJL conducted WB and animal experiments. STZ and ZJL performed RNA pulldown assay. STZ, SL, JW and GXF performed Immunofluorescence assay and. evaluation. STZ, SL, ZJL and HL interpreted and analyzed the data. STZ, SL, ZJL, YFW, XTH and XJC wrote and reviewed the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Ethical standards
Animal experiments were approved and performed in accordance with the institutional guidelines for animal care of animal ethics committee of Zhejiang University. All institutional and national guidelines for the care and use of laboratory animals were followed.
Human and animal rights
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964 and later versions.
Informed consent
Informed consent to be included in the study, or the equivalent, was obtained from all patients.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure S1
eIF4EBP3 is expressed at low levels in gastric cancer cell lines in CCLE analysis. (a) eIF4EBP3 expression was low in stomach cancer cell lines (shown in grey frame) among a variety of cancer cell lines. (b) eIF4EBP3 mRNA expression in 37 gastric cancer cell lines. (c) CpG methylation status of eIF4EBP3 in 37 gastric cancer cell lines. X axis displays the position of the methylation data, and y axis indicates the cell line in which methylation was measured. Bubble size represents coverage, and bubble color represents methylation, with warmer colors being more methylated (TIFF 2842 kb)
Figure S2
Proteomic analysis results. (a–b) Clustering heatmap and GO analysis of the significantly expressed proteins in SGC7901-eIF4EBP3 cells compared with SGC7901-vector cells. (c) β-catenin mRNA expression was upregulated in GC compared with normal liver tissue in TCGA database. (d–e) Enrichment analysis of KEGG and GO with β-catenin expression is shown. (f) PPI network of β-catenin (TIFF 1341 kb)
Rights and permissions
About this article

Cite this article
Zhai, S., Lin, S., Lin, Z. et al. eIF4EBP3 was downregulated by methylation and acted as a tumor suppressor by targeting eIF4E/β-catenin in gastric cancer. Gastric Cancer 23, 483–496 (2020). https://doi.org/10.1007/s10120-019-01030-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-019-01030-x