
Abstract
Background
Jumonji domain-containing protein 2A (JMJD2A) of the JMJD2 family of histone lysine demethylases has been implicated in tumorigenesis. However, its expression and role in gastric cancer (GC) drug resistance remain unknown. Here, we investigated the role of JMJD2A in GC chemotherapeutic susceptibility and its clinical relevance in GC.
Methods
We selected 12 relevant genes from previously identified gene signatures that can predict GC susceptibility to docetaxel, cisplatin, and S-1 (DCS) therapy. Each gene was knocked down using siRNA in GC cell lines, and cell viability assays were performed. JMJD2A expression in GC cell lines and tissues was assessed using qRT-PCR and immunohistochemistry, respectively. A JMJD2A downstream target related to drug susceptibility was examined using whole-gene expression array and immunoprecipitation.
Results
Among the 12 candidate genes, down-regulation of JMJD2A showed the maximum effect on GC susceptibility to anti-cancer drugs and increased the IC50 values for 5-FU, cisplatin, and docetaxel 15.3-, 2.7-, and 4.0-fold, respectively. JMJD2A was universally expressed in 12 GC cell lines, and its overexpression in GC tissue was positively correlated with tumor regression in 34 DCS-treated patients. A whole-gene expression array of JMJD2A-knockdown GC cells demonstrated a significant decrease in the expression of pro-apoptotic coiled-coil domain containing 8 (CCDC8), a downstream target of JMJD2A. Direct interaction between CCDC8 and JMJD2A was verified using immunoprecipitation. CCDC8 inhibition restored drug resistance to docetaxel, cisplatin, and S-1.
Conclusions
Our results indicate that JMJD2A is a novel epigenetic factor affecting GC chemotherapeutic susceptibility, and JMJD2A/CCDC8 is a potential GC therapeutic target.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Histone lysine methylation is recognized as an important post-translational modification that regulates chromatin structure and gene expression [1]. Histone demethylases remove methyl groups from the lysine residues of histone tails, thereby regulating the transcriptional activity of target genes [2, 3]. Jumonji domain-containing protein 2A (JMJD2A), also known as lysine-specific demethylase 4A (KDM4A), is a member of the histone demethylase JMJD2 (JMJD2A-2D) family that is characterized by the catalytic Jumonji C (JmjC) domain [4]. It catalyzes the demethylation of di- and tri-methylated H3K9, H3K36, and H1.4K26 [5].
Recently, the JMJD2 family has attracted much attention, because JMJD2A, JMJD2B, and/or JMJD2C have been shown to be overexpressed in breast [6], lung [7, 8], colorectal [9], and bladder tumors [10], and required for cancer cell growth [4]. In particular, JMJD2A in human gastric cancer (GC) has been identified as an oncogenic protein based on transcription regulation [11].
GC is the second leading cause of cancer-related death worldwide [12]. Recent advances in diagnostic methods and perioperative management have facilitated the early detection of GC, hence lowering the mortality rate. However, patients with advanced tumor frequently develop recurrence after radical resections, and consequently show poor survival rates [13]. Until now, many chemotherapeutic agents, with promising activity against gastric cancer, have been identified, including S-1, docetaxel, cisplatin; however, most chemotherapy regimens remain insufficient for the improvement of survival rates [14, 15]. Recently, we have shown that triple-drug combination chemotherapy comprising the key therapeutic agents for inoperable metastatic gastric cancer: docetaxel, cisplatin, and S-1 (DCS), has a high response (79.1–87.1%) and conversion surgery (22.6–34.9%) rates in patients with metastatic GC [16,17,18,19,20,21]. In addition, a phase II trial by Koizumi et al. also indicated a similarly high response rate (81%) [22]. A phase II study of neoadjuvant chemotherapy with DCS showed a sufficiently high R0 resection rate (90.7%) and appreciable pathological response (65.9%) in patients with advanced resectable GC [23]. However, unresponsive cases still occur in DCS therapy. One of the key reasons may be drug resistance, which have prevented successful treatment in these cases [24, 25]. Several research groups are still exploring various mechanisms of drug resistance to overcome this challenge of chemotherapy in advanced GC cases.
We had previously detected 29 functional gene signatures that are associated with significant response to DCS treatment using global gene expression analysis [26]. This finding provided an important clue to uncover the unidentified mechanism of drug resistance in DCS chemotherapy. Among these genes, JMJD2A was found to be one of the functional genes affecting the efficacy of DCS therapy.
Here, to extend the existing findings, we elucidated the role of JMJD2A in drug susceptibility of DCS therapy in patients with GC, and further identified significant positive correlation of tumor reduction rate with JMJD2A expression in GC. Moreover, using whole-gene expression analysis, we detected coiled-coil domain containing 8 (CCDC8) as a novel functional target of JMJD2A.
Materials and methods
Patients
Before triple-combination chemotherapy using docetaxel, cisplatin, and S-1 (DCS), a total of 34 tissues from 34 patients, with unresectable metastatic gastric cancer in clinical stage IV, were obtained in this study. Patient characteristics are shown in Table S1. Written informed consent was obtained from all patients according to the guidelines approved by the Ethics Committee of Tokushima University Hospital (Approval number; 865). Unresectable distant metastatic disease with an M1 stage was defined according to the Japanese Classification of Gastric Carcinoma (JGCA) version14 [27]. Metastatic lesions (M1) were evaluated according to the findings of conventional examinations such as contrast computed tomography, ultrasonography, or magnetic resonance imaging. Staging laparoscopy was also performed as needed to exclude occult M1 disease in the peritoneum or other intra-abdominal sites. During DCS therapy, tumor reduction rate was classified based on tumor regression rates (TRR), following the RECIST [28].
Cell lines, culture, and chemical reagents
Mainly, human gastric cancer cell lines, MKN45 and KATO-III, were used in this study. MKN45 cell lines were obtained from Japanese Collection of Research Bioresources (JCRB) Cell Bank (Osaka, Japan). KATO-III cells were provided from the department of Medical Oncology and Hematology, Sapporo Medical University School of Medicine (Hokkaido, Japan). Ten other types of human gastric cancer cells, namely MKN1, MKN74, IM95, OCUM-1, GCIY, HGC-27, NUGC-4, AGS, HSC-39, and JRST, were obtained from the JCRB Cell Bank (Osaka, Japan). Cells were maintained in RPMI 1640 medium (Sigma-Aldrich, St. Louis, MO) containing 10% FBS (Life Technologies, Carlsbad, CA) at 37 °C in an incubator with 5% CO2. 5-FU, cisplatin, and docetaxel were obtained from Sigma-Aldrich.
siRNA and transfection
Specific double-stranded JMJD2A siRNA (Silencer Select ® s18635; Ambion, Austin, TX) were transfected into MKN45 and KATO-III cells (1.0 × 104 or 1.0 × 106) using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA) following the manufacturer’s instructions. Silencer Negative Control #1 siRNA (Ambion) was used as negative control. siRNAs for the other 13 genes and CCDC8 (s38376 and s228331) were also used. The sequences of siRNAs are shown in Table S2.
Quantitative real-time PCR (qPCR)
Total RNA was extracted using RNeasy Mini Kit (Qiagen, Hilden, Germany), and reverse transcribed into cDNA using High Capacity RNA-to-cDNA kit (Life Technologies). The probe and primers were from TaqMan gene expression assay reagents (Applied Biosystems, Foster City, CA). The following TaqMan assays were used; KDM4A (Hs00206360_m1, Applied Biosystems), CCDC8 (Hs01003458_s1), 18S (Hs99999901_s1), and GAPDH (Hs02758991). Each mRNA level was normalized with respect to those of 18S and/or GAPDH as internal control.
Plasmid construction and transfection
Expression plasmid-encoding human JMJD2A protein was constructed as follows. Human JMJD2A was excised from the pOTB7 vector (Riken BioResource Center, Tsukuba, Japan) with Bam HI and Sma I and ligated into pcDNA3.1/myc-His (Invitrogen). In MKN45 cells (1.0 × 104 or 1.0 × 106), JMJD2A expression plasmid or an empty vector (pcDNA3.1/myc-His) was transiently transfected using Lipofectamine 3000 (Invitrogen).
Cell viability assay
Cell viability was assessed by water-soluble tetrazolium salt (WST) using Cell Counting Kit-8 (Dojindo, Japan). We selected 12 genes (PCGF3, B4GALT5, EGR1, PDGFB, HECA, ANTXR2, ANXA5, CISH, PLK2, ERLIN1, JMJD2A, and FAM116A) for silencing. After the incubation of siRNA or negative control siRNA for 24 h, cell growth was assayed in 96-well plates after 48 h with the treatment of 5-FU, cisplatin, and docetaxel. Various concentrations of 5-FU (up to 500 μM), cisplatin (up to 40 μM), and docetaxel (up to 1000 nM) were used, and IC50 was subsequently calculated.
Western blotting
Cells were washed with PBS and lysed in RIPA buffer (50 mM Tris–HCl, pH 8.0, with 150 mM sodium chloride, 1.0% NP-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate; Sigma-Aldrich) containing protease inhibitors (Sigma-Aldrich). Equal amounts of sample proteins were denatured by boiling for 5 min in SDS-sample buffer containing 1% beta-mercaptoethanol. The proteins were then electrophoresed on an SDS–polyacrylamide gel and transferred onto an Immobilon-P membrane (Millipore). The membrane was blocked with 5% skim milk in Tris-buffered saline containing 0.05% Tween 20 for 1 h, and thereafter incubated with a primary antibody overnight. The primary antibodies used are as follows: anti-JMJD2A mouse monoclonal antibody (1:100; sc-271210, Santa Cruz Biotechnology, Santa Cruz, CA), anti-CCDC8 rabbit polyclonal antibody (1:250; HPA041993, Sigma-Aldrich), anti-tri-methyl-histone H3 (Lys9) rabbit monoclonal antibody (1:1000; #13969 Cell Signaling Technology, Beverly, MA), anti-di-methyl-histone H3 (Lys9) rabbit monoclonal antibody (1:1000; #4658, Cell Signaling Technology), and anti-beta-actin mouse monoclonal antibody (1:10,000; A5441, Sigma-Aldrich). Proteins were then visualized with an anti-mouse and rabbit IgG horseradish peroxidase-conjugated secondary antibody (1:15,000; GE Healthcare, UK) using an ECL Western Blotting Detection System (Amersham Pharmacia Biotech, Aylesbury, UK).
Immunohistochemistry
Immunohistochemistry was performed using anti-JMJD2A polyclonal antibody (1:50; #5328, Cell Signaling Technology), and biotinylated link antibody LSAB2 System-HRP DAB kit (Dako, Carpenteria, CA) followed by counterstaining with hematoxylin.
Expression levels were visualized and classified by combining an estimate of the percentage of immunoreactive cells (quantity score) and staining intensity (staining intensity score) with minor modification, as in the previous report [8]; this was evaluated independently by two investigators. Staining intensities were divided into three grades: no staining in any tumor cell (score 0), staining of less than 50% tumor cells with weak intensity (score 1), and staining in 50–90% tumor cells with strong intensity (score 2). For instance, 60% labeled cells were scored as staining intensity 2, showing 1.2 immunohistochemistry scores (0.6 multiplied by 2), and average immunohistochemistry scores were described.
Immunoprecipitation
Immunoprecipitation (IP) kit (# K286-25) was purchased from BioVision (Milpitas, CA) and performed following the manufacturer’s protocol. Briefly, after the transfection of JMJD2A plasmid and an empty vector (pcDNA3.1/myc-His) for 48 h in MKN45 cells, cells were lysed with RIPA buffer containing protease inhibitor cocktail provided. Cell suspension were mixed for 30 min at 4 °C and centrifuged for 2 min at 2000×g, followed by extraction of the supernatant. To the cell extracts, anti-JMJD2A polyclonal antibody (Cell Signaling Technology; #5328) and rabbit IgG (Cell Signaling Technology, #3900) as control, were added and rotated overnight at 4 °C. Protein A/G beads (25 μL) were added next, and the solution was further incubated for 1 h at 4 °C, centrifuged, and the pellet washed thrice. Samples were boiled for 5 min at 95 °C and analyzed by western blotting using JMJD2A or CCDC8 antibody. Secondary antibody was used in Clean-Blot IP Detection Reagent (Thermo Scientific, Waltham, MA).
Gene expression microarray
After the transfection of JMJD2A siRNA or negative control #1 into MKN45 cells, total RNA was extracted at 24 h using an RNeasy Mini Kit (Qiagen). Four independent samples were prepared. The concentration of RNA was measured with NanoDrop 1000 spectrophotometer (NanoDrop Technologies; Thermo Scientific), and its integrity was assessed with the Agilent 2100 Bioanalyzer and RNA 6000 LabChip Kit (Agilent Technologies, Palo Alto, CA). Whole-gene expression was determined using a whole human genome microarray (SurePrint G3 Human GE 8 × 60 K; Agilent Technologies). Briefly, total RNA was labeled with Cyanine 3-Cytidine bisphosphate by T4 ligase and hybridized to SurePrint G3 human genome microarray. Subsequently, each sample was scanned by a Microarray Scanner (G2505C; Agilent Technologies), and fluorescence signal was extracted using Feature Extraction Software (version 10.7.3.1). Raw intensity of the mRNA data was analyzed by GeneSpring GX 12 (Agilent Technologies). The complete datasets were deposited in the Gene Expression Omnibus database (accession number GSE111590).
Statistical analysis
Statistical significance of the data was analyzed using the unpaired Student’s t test, using SPSS version 24.0 (SPSS, Inc., Chicago, IL). A probability level of 0.05 was used as the criterion for significance.
Results
Change in drug susceptibility after silencing 12 genes related to DCS therapy
Previously, we had identified 29 candidate predictor genes in GC tissues from both responders and non-responders of DCS therapy [26]. In this study, to extend our observation related to drug susceptibility, we selected 12 genes among the 29 genes by a leave-one-out approach as we described previously [26]. After specific silencing of the 12 genes by siRNA, the IC50 values of 5-FU, cisplatin, and docetaxel were analyzed in MKN45 cells, as shown in Table 1. Among the 12 genes, only JMJD2A showed a significant increment of each of the IC50 values of 5-FU, cisplatin, and docetaxel compared to those in control cells (15.3, 2.7, and 4.0-fold, respectively). Thus, loss of JMJD2A was associated with an increase in IC50, implicating the role of JMJD2A in resistance to the three anti-cancer drugs.
Expression of JMJD2A in 12 types of GC cells
To investigate the gene expression profiles in various histologically typified gastric cancer, we measured the expression level of JMJD2A in 12 GC cell lines (Fig. 1a). Endogenous expression of JMJD2A was universally observed in all the cell lines measured using TaqMan qPCR, whose values were normalized against both 18S and GAPDH. Among these, we selected MKN45 cells for further experimental analysis, because it showed relatively high expression of JMJD2A and is widely used as a representative cell line derived from poorly differentiated adenocarcinoma.

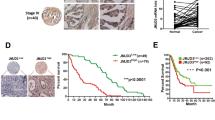
Expression of JMJD2A in human gastric cancer tissues and correlation with tumor reduction rate. a Expression levels of JMJD2A in 12 human gastric cancer cell lines were measured by TaqMan qPCR. Relative expression levels are shown as the value normalized to 18S (left) and GAPDH (right). Values are presented as means ± SD from triplicate. b Representative image showing histological staining of JMJD2A in pretreated human gastric cancer tissues; hematoxylin and eosin (HE) staining of JMJD2A (at lower (100 × ) and higher (200 × ) magnifications) (#5328). Upper panels, intestinal type (patient number 8, immunoscore of 1.8); lower panels, diffuse type (patient number 15, immunoscore of 1.7). c Correlation plots between immunohistochemistry score and tumor reduction rate (%) after two cycles of DCS therapy. p values were calculated using Pearson's test. R2 indicates the coefficient of determination
Expression of JMJD2A in human GC tissues and association with clinicopathological factors and tumor reduction rate
To examine the expression of JMJD2A and its potential clinical relevance in DCS therapy, we estimated JMJD2A expression levels in pretreatment biopsy specimens from 34 patients with GC using immunohistochemical staining. Representative histological staining of JMJD2A in intestinal type and diffuse type are shown in Fig. 1b. We first evaluated a possible association between the expression levels of JMJD2A and several clinical characteristics in patients treated with DCS. The cases in which the JMJD2A score was 0–1.0 were enrolled in the JMJD2A low group (n = 11), whereas cases with JMJD2A scores between 1.1 and 2.0 were enrolled in the JMJD2A high group (n = 23). No significant differences were observed in terms of sex, age, histologic type, HER 2 status, T stage, N stage, or distant metastasis sites (peritoneum, liver, lung, and pancreas) between the two groups, whereas significant differences in lymph node distant metastasis and a number of non-curative factors were noted (Table S3). Thereafter, we analyzed the association with reduction rates of gastric tumors. Immunoscores showed positive and significant association with JMJD2A expression after two courses of DCS therapy (p = 0.012) (Fig. 1c). Thus, high expression of JMJD2A was correlated with positive reduction rate in gastric tumors, indicating its potential role in increased drug susceptibility to DCS therapy.
Cellular function of JMJD2A
Because JMJD2A was reported to reverse tri-methylated histone H3K9, we checked the enzymatic activity of JMJD2A as a histone demethylase. We examined the loss-of and gain-of functions in JMJD2A in vitro (Fig. 2a). After the knockdown of JMJD2A in MKN45 cells, H3K9 di-methylation status was significantly reduced, and tri-methylation increased, as seen by western blotting. In contrast, with overexpressed JMJD2A, levels of H3K9 di-methylation were remarkably enhanced, and tri-methylation apparently reduced. These results were confirmed in the NUGC-4 cell line (Figure S1). This was opposite to the loss-of function result, indicating genuine methylated activity in overexpressed JMJD2A. H3K9 tri-methylation is well known as a transcriptional repressive factor to form the heterochromatin structure. In our experimental procedure, JMJD2A appeared to regulate transcription through the demethylation of H3K9.

JMJD2A physically associated with methylated histone H3K9. a JMJD2A siRNA was transfected with RNAiMAX into MKN45 (1.0 × 106) cells, and overexpressed with lipofectamine 3000. Cell extracts were measured by western blotting and evaluated by JMJD2A levels. For physiological functions of JMJD2A, cell extracts were evaluated by di- and tri-methylated H3K9 status. β-actin served as a loading control. MW; molecular weight. b Effect of JMJD2A overexpression on IC50 of 5-FU, cisplatin, and docetaxel in MKN45 cells
Drug susceptibility after overexpression of JMJD2A
Based on the loss-of-function results in JMJD2A, the IC50 values of 5-FU, cisplatin, and docetaxel were significantly increased (Table 1), suggesting the role of JMJD2A in drug resistance. We then analyzed the gain-of-function of JMJD2A to examine whether the drug susceptibility had changed. After the overexpression of JMJD2A in MKN45 cells, IC50 values of the three drugs had not changed (Fig. 2b), which was implicated in the lack of induction of drug efficacy after the overexpression of JMJD2A. In addition, using NUGC-4 cells, which showed relatively low JMJD2A expression (Fig. 1a) after overexpression of JMJD2A, we analyzed the changes of IC50 values of the three drugs (Table S4). As earlier, there was no significant modification of the IC50 values. Against the result of loss-of function in JMJD2A, we found a different status with the gain-of function of JMJD2A.
CCDC8 is a downstream target of JMJD2A
Because the interaction of JMJD2A with the other genes is unknown, we analyzed the downstream genes associated with JMJD2A silencing, using whole-gene expression analysis, to explore the mechanism by which JMJD2A regulates drug susceptibility. After silencing the JMJD2A gene in MKN45 cells, we identified 20 genes that had significantly different expression levels compared to those in control cells (unpaired t test, p < 0.05); the changes in expression of these genes were 4.5-fold greater (Fig. 3a, b). Of note, whole-gene expression analysis showed significant suppression of TXLNB, SPATA21, and CCDC8, ranked from top to third. We selected the CDCC8 gene among these candidate genes, because it is known to be a pro-apoptotic gene related to DNA damage, whereas there have been no reports describing the involvement of other genes in carcinogenesis and drug susceptibility. Moreover, we also considered that the expression of CDCC8 is reported to be relatively high in gastric cancer tissue compared to those of other genes in the TCGA database [29].

Hierarchical clustering of 20 candidate downstream genes related to JMJD2A knockdown. a Each column represents one gene, and each row represents their cellular origin. Four data from JMJD2A siRNA and four from Random siRNA in MKN45 cells, were considered, using whole human genome microarray (8 × 60 K; Agilent). A pseudo-color representation of the relative intensities is shown; red color indicates high expression, and green color low expression. b After specific gene silencing of JMJD2A, top 20 candidate genes are listed to show significant different expression
To confirm the whole-gene expression results, we used the other cellular samples and analyzed the expression of CCDC8 by qPCR and western blotting. After silencing JMJD2A in MKN45 cells, CCDC8 expression was significantly reduced at both 24 and 48 h, as shown by qPCR (Fig. 4a). In parallel, significant reduction of CCDC8 was also recognized, at 24 and 48 h, by western blot analysis (Fig. 4b).

Direct effect of JMJD2A knockdown on CCDC8 level and interaction. a After the treatment of MKN45 cells with JMJD2A siRNA, expression of JMJD2A and CCDC8 was measured at 24 h by TaqMan qPCR. Relative expression levels are shown as the value normalized to those of 18S (left) and GAPDH (right). b Levels of JMJD2A and CCDC8 were analyzed at 24 and 48 h by western blotting. β-actin was used as a control. MW: molecular weight. c MKN45 cells were transfected with expression plasmid of JMJD2A for 48 h. Transfection with empty vector was used as control. Cell lysates were immunoprecipitated with anti-JMJD2A antibody (#5328) or control IgG. The co-immunoprecipitated JMJD2A was detected by western blotting (top). The co-immunoprecipitated CCDC8 was then analyzed using anti-CCDC8 antibody (HPA041993) to evaluate the enrichment of CCDC8 (bottom). MW: molecular weight
To further explain the cellular association of CCDC8, we tested the in-vitro interaction of JMJD2A and CCDC8 using immunoprecipitation assay. As shown in Fig. 4c, CCDC8 bound to endogenous JMJD2A, but not to IgG alone, showing that CCDC8 and JMJD2A interact directly.
Effect of CCDC8 knockdown on drug susceptibility
In parallel with the loss of JMJD2A, the IC50 values of 5-FU, cisplatin, and docetaxel were found to be significantly increased (Table 1). Thus, we analyzed whether CCDC8 function was involved in drug susceptibility (Table 2). After silencing of CCDC8 (Figure S2), the IC50 value of 5-FU, cisplatin, and docetaxel were significantly higher than in control cells, indicating that CCDC8 cooperates with JMJD2A to affect the modulation of susceptibility to 5-FU, cisplatin, and docetaxel, and induced drug resistance in DCS therapy.
Discussion
In this study, we showed that JMJD2A can potentially regulate drug susceptibility in DCS therapy using siRNA-mediated down-regulation and immunohistochemistry of GC patient tissues. Additionally, using whole-gene expression analysis and in-vitro immunoprecipitation, we identified that JMJD2A cooperates with CCDC8 to control the susceptibility of 5-FU, cisplatin, and docetaxel treatment.
Aberrant changes in histone lysine methylation status have been linked to cancer formation [30]. In a recent study, members of the JMJD2 family of histone lysine demethylases (KDM) have been identified as having oncogenic function and are highly expressed in human cancers [31]. In this regard, Hu et al. reported that high JMJD2A levels are associated with disease stage, nodal status, and metastasis in GC patients [11]. Similarly, when we analyzed the relationship between expression of JMJD2A and clinicopathological factors of GC patients, significant differences were observed in distant lymph node metastasis and the number of non-curative metastatic factors (Table S3). Thus, JMJD2A has been indicated to be involved in tumorigenesis and in enhancing the metastasis of GC. However, its role in anti-cancer drug resistance in GC therapy remains largely unknown.
Histone methylation can positively or negatively impact gene transcription; dysregulation of histone methyl-modifying enzymes has been increasingly recognized as a hallmark of carcinogenesis [32, 33]. JMJD2A is a transcriptional cofactor and enzyme that catalyzes demethylation of H3K9 and H3K36 [4, 34]. Because methylation of H3K9 in promoter regions is associated with transcriptional repression [35], JMJD2A plays an important role in diverse types of cancer, including breast [6], lung [7, 8], colon [9], and bladder cancer [10], possibly activating transcription through demethylation of H3K9 [7]. For instance, overexpression of JMJD2A is sufficient to promote replication and copy gain of specific chromosomal regions, which is associated with the drug-resistant oncogene CKS1B in breast cancer [36]. Using a knockdown approach, we observed a significant contribution of JMJD2A to the susceptibility to key anti-cancer drugs (5-FU, cisplatin, and docetaxel) for treatment of GC. Our data raise the possibility that loss of JMJD2A function may result in inactivation of transcription of genes related to drug susceptibility, which leads to increase in the IC50 value of these three drugs.
Given the role of JMJD2A in gene transcription, it was worth investigating a cooperative novel factor in the regulation of drug susceptibility. One advantage of unbiased whole-gene expression approach is the potential to uncover new genes that can be targeted for further investigation. Thus, we analyzed the effects on global gene expression of JMJD2A silencing. We identified CCDC8 as the downstream target of JMJD2A, which was verified using in vitro experiments.
CCDC8 contains two coiled-coil domains [37] involved in diverse biological processes such as the regulation of gene expression, cell division, and membrane fusion [38, 39]. CCDC8 mutation leads to the development of 3 M syndrome in humans, a primordial growth disorder, by interaction with CUL7 (Cullin 7) and OBSL1 (Obscurin-Like 1) [37]. In addition, CCDC8 has been previously shown to be a tumor suppressor in several types of cancer, such as breast [40], renal [41], and lung cancers [42], associated with hypermethylation of the CCDC8 gene in some of these tumors.
However, the role and possible mechanisms of CCDC8 in drug susceptibility has not been elucidated to date. Interestingly, CCDC8 is also known as a critical cofactor for p53-mediated apoptosis upon DNA damage and as facilitating activation of proapoptotic targets [43]. In this regard, p53-mediated apoptosis is abrogated in the gastric tumor cells lacking CCDC8 expression, which leads to resistance to chemotherapeutic agents such as CDDP and 5FU that are known to cause p53-dependent apoptosis [44].
Moreover, knockdown of CCDC8 clearly induced drug tolerance against docetaxel in our analysis. This drug, belonging to the taxane family of compounds, exerts anti-tumor activity by promoting the assembly of microtubules and preventing their de-polymerization, resulting in cell apoptosis [45, 46]. The ordered assembly and disassembly of microtubules are tightly regulated by numerous microtubule-associated proteins [47]. In a recent report, deletion of CCDC8 was shown to result in microtubule defects, thus affecting fundamental cellular processes in patients with 3 M syndrome [48]. Interestingly, we found that low levels of CCDC8 expression closely associated with anti-cancer drug resistance, supporting the notion that docetaxel tolerance, mediated by disassembly of microtubules, is caused by loss of CCDC8. However, the detailed mechanism underlying drug susceptibility related to CCDC8 remains unclear. Further research is required to clarify how CCDC8 affects chemotherapeutic drug susceptibility in relation to JMJD2A in GC.
In our experimental settings, overexpression of JMJD2A did not show the same phenotype in chemosensitivity (Fig. 2b, Table S4), although loss of JMJD2A affected drug susceptibility via lack of CCDC8. Genetic studies have shown that deletion of histone demethylases lead to pleiotropic phenotypes. In general, deletion or mutation of histone demethylases has been shown to induce severe phenotypes. For instance, LSD1 (also known as KDM1A) knockout mice display embryonic lethality around E5.5) [49]. In contrast, most studies have revealed the effects of histone demethylases on transcription of a limited number of genes [32]. These studies show that demethylases bind to a large number of genes, but the expression of only a limited number of genes was affected. Thus, the effect of histone demethylases is supposed to be generally modest. In fact, CCDC8 levels were not increased after overexpression of JMJD2A (Fig. 4c), suggesting that JMJD2A may not fully interact with methylation residues of the CCDC8 gene promoter. This may partly explain why overexpression of JMJD2A did not enhance the chemotherapeutic drug susceptibility of GC cells.
On the contrary, a positive correlation of JMJD2A expression and tumor reduction rate was observed in DCS therapy (Fig. 1c), indicating that JMJD2A may sensitize GC to chemotherapeutic drugs. Although the underlying mechanisms of these inconsistent results between in vitro and in vivo functions of JMJD2A were not fully investigated in the present study, a direct interaction between JMJD2A and CCDC8 as evidenced by immunoprecipitation assays (Fig. 4c) suggests that JMJD2A may exert its drug susceptibility function in cooperation with CCDC8. The different mechanisms of this cooperation may depend on the biological environment, such as specific methylation status in vivo and in vitro. To develop therapies targeting JMJD2A/CCDC8, further research is required to unveil the mechanism by which JMJD2A/CCDC8 interaction modulates anticancer drug susceptibility.
In conclusion, our study revealed that JMJD2A is a novel epigenetic factor of chemotherapeutic susceptibility in GC, and high expression of JMJD2A was found to reliably predict tumor regression rates with DCS therapy, thereby showing that JMJD2A could serve as an independent factor in the treatment with triple anti-cancer drugs. Using a whole-gene expression approach, we identified CCDC8 as the direct downstream gene of JMJD2A and demonstrated the cooperation between JMJD2A and CCDC8 in the regulation of anti-cancer drugs, which may be a promising molecular target for the treatment of GC.
References
Chi P, Allis CD, Wang GG. Covalent histone modifications-miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–69.
Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–12.
Shi Y, Whetstine JR. Dynamic regulation of histone lysine methylation by demethylases. Mol Cell. 2007;25:1–14.
Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. 2013;73:2936–42.
Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012;48:491–507.
Berry WL, Shin S, Lightfoot SA, Janknecht R. Oncogenic features of the JMJD2A histone demethylase in breast cancer. Int J Oncol. 2012;41:1701–6.
Kogure M, Takawa M, Cho H-S, Toyokawa G, Hayashi K, Tsunoda T, et al. Deregulation of the histone demethylase JMJD2A is involved in human carcinogenesis through regulation of the G(1)/S transition. Cancer Lett. 2013;336:76–84.
Mallette FA, Richard S. JMJD2A promotes cellular transformation by blocking cellular senescence through transcriptional repression of the tumor suppressor CHD5. Cell Rep. 2012;2:1233–43.
Kim T-D, Shin S, Berry WL, Oh S, Janknecht R. The JMJD2A demethylase regulates apoptosis and proliferation in colon cancer cells. J Cell Biochem. 2012;113:1368–76.
Kauffman EC, Robinson BD, Downes MJ, Powell LG, Lee MM, Scherr DS, et al. Role of androgen receptor and associated lysine-demethylase coregulators, LSD1 and JMJD2A, in localized and advanced human bladder cancer. Mol Carcinog. 2011;50:931–44.
Hu C-E, Liu Y-C, Zhang H-D, Huang G-J. JMJD2A predicts prognosis and regulates cell growth in human gastric cancer. Biochem Biophys Res Comm. 2014;449:1–7.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin. 2018;68:394–424.
Ajani JA, Lee J, Sano T, Janjigian YY, Fan D, Song S. Gastric adenocarcinoma. Nat. Rev. Dis. Primers. 2017;3:17036–119.
Shen L, Shan Y-S, Hu H-M, Price TJ, Sirohi B, Yeh K-H, et al. Management of gastric cancer in Asia: resource-stratified guidelines. Lancet Oncol. 2013;e535–47.
Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet. 2016;388:2654–64.
Takayama T, Sato Y, Sagawa T, Okamoto T, Nagashima H, Takahashi Y, et al. Phase I study of S-1, docetaxel and cisplatin combination chemotherapy in patients with unresectable metastatic gastric cancer. Br J Cancer. 2007.
Sato Y, Takayama T, Sagawa T, Takahashi Y, Ohnuma H, Okubo S, et al. Phase II study of S-1, docetaxel and cisplatin combination chemotherapy in patients with unresectable metastatic gastric cancer. Cancer Chemother Pharmacol. 2009;66:721–8.
Mitsui Y, Sato Y, Miyamoto H, Fujino Y, Takaoka T, Miyoshi J, et al. Trastuzumab in combination with docetaxel/cisplatin/S-1 (DCS) for patients with HER2-positive metastatic gastric cancer: feasibility and preliminary efficacy. Cancer Chemother Pharmacol. 2015;76:375–82.
Uemura N, Kikuchi S, Sato Y, Ohnuma H, Okamoto K, Miyamoto H, et al. A phase II study of modified docetaxel, cisplatin, and S-1 (mDCS) chemotherapy for unresectable advanced gastric cancer. Cancer Chemother Pharmacol. 2017;80:707–13.
Sato Y, Ohnuma H, Nobuoka T, Hirakawa M, Sagawa T, Fujikawa K, et al. Conversion therapy for inoperable advanced gastric cancer patients by docetaxel, cisplatin, and S-1 (DCS) chemotherapy: a multi-institutional retrospective study. Gastric Cancer. 2017;20:517–26.
Ohnuma H, Sato Y, Hirakawa M, et al. Docetaxel, cisplatin and S-1 (DCS) combination chemotherapy for gastric cancer patients with peritoneal metastasis: a retrospective study. Cancer Chemother Pharmacol. 2018;81(3):539–48.
Koizumi W, Nakayama N, Tanabe S, Sasaki T, Higuchi K, Nishimura K, et al. A multicenter phase II study of combined chemotherapy with docetaxel, cisplatin, and S-1 in patients with unresectable or recurrent gastric cancer (KDOG 0601). Cancer Chemother Pharmacol. 2011;69:407–13.
Hirakawa M, Sato Y, Ohnuma H, Takayama T, Sagawa T, Nobuoka T, et al. A phase II study of neoadjuvant combination chemotherapy with docetaxel, cisplatin, and S-1 for locally advanced resectable gastric cancer: nucleotide excision repair (NER) as potential chemoresistance marker. Cancer Chemother Pharmacol. 2013;71:789–97.
Pietrantonio F, De Braud F, Da Prat V, Perrone F, Pierotti MA, Gariboldi M, et al. A review on biomarkers for prediction of treatment outcome in gastric cancer. Anticancer Res. 2013;33:1257–66.
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26.
Kitamura S, Tanahashi T, Aoyagi E, Nakagawa T, Okamoto K, Kimura T, et al. Response predictors of S-1, cisplatin, and docetaxel combination chemotherapy for metastatic gastric cancer: microarray analysis of whole human genes. Oncology. 2017;93:127–35.
Japanese Gastric Cancer Association. Japanese classification of gastric carcinoma: 3rd English edition. Gastric Cancer. 2011. pp. 101–12.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;228–47.
Human Protein Atlas available from https://www.proteinatlas.org
Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat Rev Genet. 2007;8:829–33.
Cloos PAC, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, et al. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–11.
Kooistra SM, Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol. 2012;13:297–311.
Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–57.
Guerra-Calderas L, González-Barrios R, Herrera LA, Cantú de León D, Soto-Reyes E. The role of the histone demethylase KDM4A in cancer. Cancer Genet. 2015;208:215–24.
Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–37.
Black JC, Zhang H, Kim J, Getz G, Whetstine JR. Regulation of transient site-specific copy gain by MicroRNA. J Biol Chem. 2016;291:4862–71.
Hanson D, Murray PG, O'Sullivan J, Urquhart J, Daly S, Bhaskar SS, et al. Exome sequencing identifies CCDC8 mutations in 3-M syndrome, suggesting that CCDC8 contributes in a pathway with CUL7 and OBSL1 to control human growth. Am J Hum Genet. 2011;89:148–53.
Burkhard P, Stetefeld J, Strelkov SV. Coiled coils: a highly versatile protein folding motif. Trends Cell Biol. 2001;11:82–8.
Woolfson DN. The design of coiled-coil structures and assemblies. Adv Protein Chem. 2005;70:79–112.
Pangeni RP, Channathodiyil P, Huen DS, Eagles LW, Johal BK, Pasha D, et al. The GALNT9, BNC1 and CCDC8 genes are frequently epigenetically dysregulated in breast tumours that metastasise to the brain. Clin Epigenet. 2015;7:57.
Morris MR, Ricketts CJ, Gentle D, McRonald F, Carli N, Khalili H, et al. Genome-wide methylation analysis identifies epigenetically inactivated candidate tumour suppressor genes in renal cell carcinoma. Oncogene. 2011;30:1390–401.
Jiang G-Y, Zhang X-P, Zhang Y, Xu H-T, Wang L, Li Q-C, et al. Coiled-coil domain-containing protein 8 inhibits the invasiveness and migration of non-small cell lung cancer cells. Hum Pathol. 2016;56:64–73.
Dai C, Tang Y, Jung SY, Qin J, Aaronson SA, Gu W. Differential effects on p53-mediated cell cycle arrest vs. apoptosis by p90. Proc Natl Acad Sci USA. 2011;108:18937–42.
Matsuhashi N, Saio M, Matsuo A, Sugiyama Y, Saji S. The evaluation of gastric cancer sensitivity to 5-FU/CDDP in terms of induction of apoptosis: time- and p53 expression-dependency of anti-cancer drugs. Oncol Rep. 2005;14:609–15.
Cortes JE, Pazdur R. Docetaxel. J Clin Oncol. 1995;13:2643–55.
Bissery MC, Guénard D, Guéritte-Voegelein F, Lavelle F. Experimental antitumor activity of taxotere (RP 56976, NSC 628503), a taxol analogue. Cancer Res. 1991;51:4845–52.
Glotzer M. The 3Ms of central spindle assembly: microtubules, motors and MAPs. Nat Rev Mol Cell Biol. 2009;10:9–20.
Yan J, Yan F, Li Z, Sinnott B, Cappell KM, Yu Y, et al. The 3M complex maintains microtubule and genome integrity. Mol Cell. 2014;54:791–804.
Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009;41:125–9.
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI [Grant number JP16K19348 and JP18K07942]. We thank Hideaki Horikawa for analysis of microarray data, supported by the Support Center for Advanced Medical Sciences, Tokushima University Graduate School of Biomedical Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Tetsuji Takayama received a research fund from TAIHO Pharmaceutical Co., Ltd. (Tokyo, Japan).
Human rights statement
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964 and later versions.
Informed consent
Informed consent or a substitute for it was obtained from all patients for being included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nakagawa, T., Sato, Y., Tanahashi, T. et al. JMJD2A sensitizes gastric cancer to chemotherapy by cooperating with CCDC8. Gastric Cancer 23, 426–436 (2020). https://doi.org/10.1007/s10120-019-01024-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-019-01024-9