
Abstract
Background
We conducted a multicenter phase II trial to assess the suitability of three types of chemotherapy (docetaxel plus S-1, irinotecan plus S-1, or S-1 alone) for patients with advanced gastric cancer by means of the collagen gel droplet embedded culture-drug sensitivity test (CD-DST). To our knowledge, this is the first multicenter clinical trial that has employed CD-DST to choose anticancer agents for the treatment of advanced gastric cancer.
Methods
Subjects (n = 64) were patients with advanced or recurrent gastric cancer. Patients were allocated to one of the treatment regimens on the basis of CD-DST results. Outcome of the patients was compared between the groups deemed chemosensitive or chemoresistant by the CD-DST.
Results
Thirty-three patients showed high sensitivity (T/C ratio <60 %) to at least one type of anticancer agent (sensitive group), and 31 showed low sensitivity (T/C ratio ≥60 %) to all agents (resistant group). Specifically, the 1-year survival rate was significantly higher in the sensitive group (78.5 %; 95 % CI, 67.2–94.7 %) than in the resistant group (54.7 %; 95 % CI, 38.7–74.3 %; P = 0.019), whereas time to progression (TTP) was significantly longer in the sensitive group (59.8 %; 95 % CI, 48.2–81.7 %) than in the resistant group (30.0 %; 95 % CI 13.6–46.4 %; P = 0.023). Median survival time was also significantly longer in the sensitive group (15.5 months; 95 % CI, 12.8–18.2) than in the resistant group (12.5 months; 95 % CI, 10.2–14.9; P = 0.038).
Conclusions
CD-DST predicts the outcome of patients undergoing chemotherapy for advanced gastric cancer, presumably through evaluating chemosensitivity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The prognosis of patients with resectable gastric cancer has improved with the development of technologies that enable early diagnosis and progress in surgical technique and perioperative management. However, prognosis remains extremely poor for those with locally advanced or recurrent cases or those with distant metastasis. Several anticancer agents were recently introduced and have boosted the hope of better chemotherapy outcome. A regimen most commonly used globally both in the clinical practice and as a reference arm in phase III trials had been a combination of cisplatin (CDDP) and 5-fluorouracil (5-FU) [1–3]. Given the approval of several types of new drugs, however, we hypothesized that personalized therapy guided by adequate chemosensitivity testing could lead to superior outcome when compared with the empirical therapy. For this purpose, the collagen gel droplet embedded culture-drug sensitivity test (CD-DST), a new and sophisticated method, was expected to serve an important role. Thus, we organized the Shiga Clinical Study Group for Chemosensitivity Tests for Gastrointestinal Cancer, which comprises 20 participating institutions, and conducted a stratified phase II trial for the combination treatment of docetaxel (TXT) and S-1, irinotecan (CPT-11) and S-1, or S-1 alone in advanced gastric cancer guided by CD-DST. Participants were patients with advanced or recurrent gastric cancer who were treated with anticancer drugs according to CD-DST chemosensitivity results for TXT, CPT-11, and 5-FU.
Materials and methods
Objective
The objective of this study was to prove that CD-DST predicts survival of patients who undergo chemotherapy for advanced gastric cancer: that 1-year survival rate of patients who were determined as chemosensitive by CD-DST is significantly higher than that of those determined as chemoresistant.
Eligibility
Patients with recurrent or advanced gastric cancer who suffer from either unresectable or residual disease were eligible. Availability of fresh tissue samples for CD-DST was also a prerequisite for enrollment. Gastrectomy in the current study was therefore performed either to palliate symptoms related to the primary lesion or as a reduction surgery. In addition, patients whose metastases were found during surgery were also eligible, provided the fresh samples could be harvested and transported immediately for CD-DST. Patients with unresectable primary lesion were eligible only when sufficient biopsy samples were available for chemosensitivity testing. Patients with recurrent disease were also eligible when fresh specimens of the recurrent cancer were available. Other inclusion criteria were as follows: age of 20–79 years; histologically proven gastric cancer; no previous chemotherapy and/or radiotherapy with the exception of adjuvant chemotherapy given after curative surgery. ECOG performance status score of 0–1; capable of oral ingestion; predicted survival of 3 months or more from the first day of chemotherapy; satisfactory function of bone marrow, heart, liver, and kidney; and ability to provide written consent. Between August 2007 and May 2009, 80 patients from 20 medical institutes in Shiga Prefecture, Japan, were enrolled in the study. The study was formally approved by the ethics committee of each participating institute and conducted in accordance with the principles of the Declaration of Helsinki. Written informed consent was obtained from all patients.
CD-DST procedure
Immediately after surgical resection of the tumor in each patient, a viable portion of the tumor was identified and resected by a physician who was not involved in the surgery itself to avoid delaying the surgery. The resected tumor was immediately stored in culture medium at 4 °C, and CD-DST was started promptly on the same day as the surgery. A single operator (K.I.) performed all CD-DST assays at a laboratory in Shiga University of Medical Science to evaluate sensitivities to TXT, CPT-11 (SN-38), and 5-FU. The CD-DST procedure was performed similarly with the biopsy specimens when samples weighing 0.1–0.5 g in total were available.
CD-DST was carried out according to the method reported by Kobayashi et al. [4, 5], who invented the method using a human tumor cell primary culture system kit (Primaster®; Kurabo Industries Ltd., Osaka, Japan). Briefly, each sample was washed five times with 50 ml saline containing 1.0 mg/ml penicillin, 0.5 mg/ml kanamycin, and 2.5 μg/ml amphotericin B and treated afterwards with Dispersion Enzyme Cocktail EZ (Primaster® reagent). Obtained cell suspension samples were inoculated into collagen gel-coated flasks (CG flasks, a Primaster® device) and cultured overnight in pre-culture medium PCM-1 (Primaster® content) at 37 °C in 5 % CO2. Next, the collagen gel was digested with 0.05 % EZ, and viable cancer cells were obtained. Type I collagen, 10× concentrated F-12 medium, and reconstitution buffer were mixed together in ice water with a ratio of 8:1:1 (Primaster® content). The prepared cancer cell suspension was added to the collagen solution at a final density of 1 × 105 cells/ml. Three drops of the collagen-cell mixture (30 μl/drop) were placed in each well of a 6-well plate on ice and allowed to gel at 37 °C in a CO2 incubator; the final concentration was about 3 × 103 cells per collagen gel droplet. DF medium containing 10 % fetal bovine serum (FBS) was overlaid in each well 1 h later, and plates were incubated overnight in a CO2 incubator at 37 °C. The anticancer drugs were added at the following final concentrations and incubated for 24 h: 0.1 μg/ml TXT, 0.03 μg/ml CPT-11 (SN-38), and 1.0 μg/ml 5-FU. The concentration of each anticancer drug in the culture medium was determined so as to exhibit the same area under the curve value as observed in the serum during the first 24 h after the intravenous administration of the corresponding drug at the standard clinical dosage.
After removal of the medium containing the anticancer drugs, each well was rinsed twice with 3 ml Hanks’ balanced salt solution, overlaid with 4 ml PCM-2 medium (Primaster® serum-free medium), and incubated for a further 7 days. At the end of the incubation, a neutral red solution was added to each well at a final concentration of 50 μg/ml, and colonies in the collagen gel droplets were stained for 2 h. Each collagen droplet was fixed with 10 % neutral buffered formalin, washed in water, air dried, and quantified by optical density image analysis using the Primage System (Solution Systems, Tokyo, Japan). Samples with an optical density >3.0 in the control wells were regarded as evaluable samples. In vitro sensitivity was expressed as the T/C ratio, where T is the optical density of the treated samples and C is the optical density of the controls; a T/C ratio <60 % was regarded as chemosensitive in vitro. The cutoff value at 60 % was used in the current clinical trial because the percentage of patients determined as chemosensitive had been 29.6, 28.6, and 47.3 % for 5-FU, CPT-11, and TXT, respectively, among 30 samples tested by the same investigators in a preparatory pilot study. These percentages were relatively close to the response rates of each drug in the clinical setting.
Study design, patient allocation, and treatments
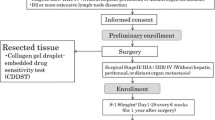
We hypothesized that therapy with anticancer drugs to which patients were deemed sensitive would be more effective than therapy with anticancer drugs that were blindly selected. Based on this hypothesis, we opted for a nonrandomized method where patients were allocated to personalized anticancer drugs predetermined by CD-DST. Patients were allocated to one of the following three treatment regimens: TXT/S-1 (TXT), CPT-11/S-1 (CPT), or S-1 (S-1; Fig. 1). Briefly, when CD-DST results showed sensitivity to all three anticancer drugs, patients were allocated to the regimen with the drug predicted to be most effective, that is, the drug with the lowest T/C ratio (sensitive group). When CD-DST results showed sensitivity to either TXT or CPT-11, or only to S-1, patients were allocated to the regimen with the corresponding drug (sensitive group). When cancer cells were not sensitive to any of the drugs, patients were randomly allocated (resistant group). Figure 2 shows details of the TXT, CPT-11, and S-1 regimens.

Allocation of chemotherapy on the basis of collagen gel droplet embedded culture-drug sensitivity test (CD-DST) results

Chemotherapy schedule
Treatment was discontinued in the event of serious adverse events, disease progression, or patient refusal, or when the physician in charge decided that the treatment should be discontinued. Further lines of treatment were to be given at the discretion of the physicians.
In the aforementioned pilot study with a cutoff value of T/C ratio at 60 %, 8 of 30 samples (26.4 %) were deemed chemoresistant to all three drugs. The difference in 1-year survival rate between these patients and 16 patients who were determined as chemosensitive was 25 % (the chemosensitivity test failed in the other 6 patients). To detect a similar difference in 1-year survival rate at α = 0.05 and β = 0.2, the sample size of the study was calculated to be 144 patients, of whom 38 patients were expected to be rated as chemoresistant.
Patient evaluation
The primary endpoint was 1-year survival rate. The secondary endpoints were time to progression (TTP), median survival time (MST), and response rate. The response was evaluated in accordance with the Response Evaluation Criteria in Solid Tumors (RECIST). Acute toxicity was graded according to National Cancer Institute Common Toxicity ver. 3.0.
Statistical analysis
Survival curves were calculated according to the Kaplan–Meier method. A generalized Wilcoxon test was used to determine significant differences between curves. The chi-squared test and Student’s t test were used to determine differences between groups. P < 0.05 was considered statistically significant.
Results
Patient accrual
Results of pivotal phase III trials emerged while our trial was ongoing. A regimen of CDDP plus S-1 have shown significant improvement in overall survival over S-1 monotherapy in a randomized comparison for advanced gastric cancer (SPIRITS trial) [6]. More recently, a regimen consisting of capecitabine, CDDP, and trastuzumab whose efficacy was proven in the ToGA trial became a standard chemotherapy for HER2-positive advanced gastric cancer [7]. Because results of the SPIRITS trial were promptly reflected in the Japanese Gastric Cancer Treatment Guidelines 2010 and a combination of S-1 and CDDP became the standard of care, the Shiga Clinical Study Group for Chemosensitivity Tests for Gastrointestinal Cancer decided that offering other regimens as a first-line therapy constituted an infringement of ethics. The final decision was that the trial should evaluate 80 patients who had already completed the 1-year follow-up at the time of decision (total number of patients recruited at the time was 129). Of these 80 patients, CD-DST results were successfully obtained in 64 patients (80 %). Failure to obtain results in the remaining 16 patients (20 %) was mainly the result of contamination from bacterial infection. Thus, 64 patients with CD-DST results were further assessed (Table 1).
Patient characteristics
The sensitive group comprised 33 patients and the resistant group comprised 31 patients. Characteristics, allocated regimens, and cycles of chemotherapy in both groups are shown in Table 1. Because the chemotherapeutic regimen in the sensitive group was allocated on the basis of sensitivity results whereas the allocation in the resistant group was random, the number of patients undergoing S-1 monotherapy turned out to be significantly smaller in the sensitive group than in the resistant group.
Efficacy
No significant difference in survival was noted among the three regimens (Fig. 3a). The 1-year survival rate was significantly higher in the sensitive group (78.5 %; 95 % CI, 67.2–94.7 %) than in the resistant group (54.7 %; 95 % CI, 38.7–74.3 %; P = 0.019; Fig. 3b). TTP was significantly longer in the sensitive group (59.8 %; 95 % CI, 48.2–81.7 %) than in the resistant group (30.0 %; 95 % CI, 13.6–46.4 %; P = 0.023; Fig. 3c). MST was also significantly longer in the sensitive group (15.5 months; 95 % CI, 12.8–18.2) than in the resistant group (12.5 months; 95 % CI, 10.2–14.9; P = 0.038).

Survival curve on the basis of chemotherapy regimen. a No significant difference in survival was noted between the three regimens. Survival curve on the basis of CD-DST sensitivity. b The survival rate was significantly higher in the sensitive group than in the resistant group. Time to progression on the basis of CD-DST sensitivity. c TTP was significantly longer in the sensitive group than in the resistant group. Survival curve on the basis of CD-DST sensitivity except for S-1. d The sensitive group showed significantly better survival than the resistant group
Because the number of patients undergoing the S-1 regimen was significantly lower in the sensitive group than in the resistant group, we reevaluated the survival curves after excluding patients who were treated by S-1 alone from both groups. In this analysis, the sensitive group showed significantly better survival (79.0 %; 95 % CI, 68.5–96.4 %) than the resistant group (62.0 %; 95 % CI, 44.1–85.9 %; Fig. 3d; P = 0.043).
The confirmed disease response rate was 45.4 % in the sensitive group (95 % CI, 20.3–73.1 %) and 32.3 % in the resistant group (95 % CI, 7.2–67.7 %). There were no statistical differences (P = 0.450; Table 2 ).
Toxicity
Adverse events are summarized in Table 3. The incidence of adverse events tended to be lower in patients who underwent the S-1 regimen, but no statistically significant difference was found among the three arms. The incidence of adverse events with toxicity grade 3 or more in the patients who underwent the TXT regimen was comparable to that in patients who underwent the CPT-11 regimen. Temporary discontinuation of chemotherapy was necessary in some cases, but there were no treatment-related deaths.
Discussion
The efficacy of molecularly targeted drugs is often predicted by analyzing protein or gene expression. For example, HER2 expression is a predictor of the efficacy of trastuzumab [7], whereas epidermal growth factor receptor (EGFR) expression and the mutation status of the K-Ras gene have been shown to be predictors of cetuximab and panitumumab efficacy [8, 9]. In contrast, pursuit of biomarkers that reflect chemosensitivity of cytotoxic agents had been more problematic, and the in vitro chemosensitivity test remains a practical option in prediction of response to chemotherapy. CD-DST is a chemosensitivity test wherein isolated tumor cells are embedded in collagen droplets. This three-dimensional culture system provides CD-DST with the following advantages over conventional methods: small specimens can be used; the effect of anticancer drugs at physiological concentrations can be assessed; and the masking effect caused by fibroblast contaminations in culture can be eliminated (with the aid of an image analysis system). Consequently, the system provides results that reflect only the anticancer drug effect on cancer cells [4, 5]. Although CD-DST was used to assess sensitivity to S-1, TXT, and CPT-11 in the present study, any anticancer agents can be tested by this method. It may be useful to predict the efficiency of CDDP/5-FU combination chemotherapy, a regimen commonly used worldwide.
The efficacy of CD-DST in cancer treatment has previously been demonstrated [10–13]. CD-DST also proved useful in chemotherapy for residual or recurrent non-small cell lung cancer [14] and for predicting the effect of preoperative chemotherapy for tumor size reduction in patients with advanced or recurrent breast cancer [15]. When outcomes of adjuvant chemotherapy with S-1 were examined in gastric cancer patients, 3-year survival rates and relapse-free survival rates were significantly higher in those with high chemosensitivity [16]. However, to the best of our knowledge, the efficacy of CD-DST in the treatment of advanced gastric cancer has not been studied previously.
We found that 1-year survival rates, TTP, and MST were significantly improved when treated with drugs predicted by CD-DST to be sensitive. Because of the small number of patients, our results may not be sufficient to claim the efficacy of CD-DST in gastric cancer treatment, but they do indicate the potential for CD-DST in selecting anticancer drugs for use in personalized medicine. One drawback is a possibility that the inferior survival time of the resistant group merely reflects more aggressive biology, theoretically associated with inclusion of greater proportion of cancer stem cells or cancer cells that have undergone epithelial to mesenchymal transition. A larger trial with more sophisticated design is needed to overcome this argument.
S-1 is the most frequently used type of fluoropyrimidines for treating gastric cancer patients in Japan. TXT and CPT-11 are also commonly used. When we embarked on this study, TXT/S-1 and CPT-11/S-1 had been regarded as promising candidates for standard first-line chemotherapy for gastric cancer and had actually been under evaluation in phase III trials with S-1 monotherapy as a control. It was only after a series of phase III trials that S-1/CDDP became the standard first-line treatment in Japan. In non-Asian countries, combinations such as infusional 5-FU/CDDP, capecitabine/CDDP, and capecitabine/oxaliplatin have been frequently prescribed for treating advanced gastric cancer. S-1/CDDP was also approved in 30 European countries after the favorable safety profile was revealed in the FLAGS trial [3]. Thus, a combination of oral or infusional fluoropyrimidine with a platinum agent can be considered as the current standard of care for gastric cancer worldwide. In the current study, no significant difference in survival was noted among the three regimens, and no comparison was made between these and S-1/CDDP. However, the main purpose of this study was to examine the relevance of CD-DST in the treatment of advance gastric cancer and not to make comparisons in efficacy between various treatment regimens. Our results suggested that a personalized therapy guided by adequate chemosensitivity testing could lead to superior outcome when compared with the standard treatment. To robustly clarify this issue, a larger trial as proposed by Schrag et al. [17] might be necessary. In that trial, patients in the exploratory arm receive one of several treatments including S-1/CDDP based on the chemosensitivity test while all patients in the control group receive S-1/CDDP.
Despite the introduction of several new promising anticancer drugs, none appears to produce satisfactory outcomes in patients. S-1 was shown to significantly improve the survival rate of gastric cancer patients when used as adjuvant chemotherapy after gastrectomy [18]. However, S-1 as a monotherapy was merely noninferior to the conventional infusional 5-FU in the treatment of advanced gastric cancer. Meanwhile, phase III studies comparing single agent (S-1 alone) and its combination with newly approved drugs (TXT plus S-1 or CPT-11 plus S-1) eventually showed no difference in survival rates [19, 20]. In the present study, the 1-year survival rate in both combination chemotherapy regimens was higher in the sensitive group in terms of CD-DST than in the resistant group. If CD-DST had been employed in the aforementioned phase III studies, a significant survival advantage might have been observed in the patients who were shown to be chemosensitive for TXT or CPT-11. The notion of incorporating chemosensitivity testing with a relevant phase III trial as proposed by Wieand [21] may thus be another means of validating the concept of the chemosensitivity assay. According to his concept, investigating CD-DST results in both responder and nonresponder subgroups in future phase III trials of anticancer drugs will confirm the clinical significance of CD-DST in chemotherapy.
To conclude, a superior 1-year survival rate was observed among chemosensitive patients who received CD-DST-guided treatment when compared with chemoresistant patients for whom the treatment was randomly allocated. Thus, CD-DST might be helpful for selecting appropriate anticancer drugs in the treatment of advanced gastric cancer.
References
Van Cutsem E, Moiseyenko VM, Tjulandin S, Majlis A, Constenla M, Boni C, et al. Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol. 2006;24:4991–7.
Kang YK, Kang WK, Shin DB, Chen J, Xiong J, Wang J, et al. Capecitabine/cisplatin versus 5-fluorouracil/cisplatin as first-line therapy in patients with advanced gastric cancer: a randomised phase III noninferiority trial. Ann Oncol. 2009;20:666–73.
Ajani JA, Rodriguez W, Bodoky G, Moiseyenko V, Lichinitser M, Gorbunova I, et al. Multicenter phase III comparison of cisplatin/S-1 with cisplatin/infusional Fluorouracil in Advanced Gastric or Gastroesopageal Adenocarcinoma Study: the FLAGS Trial. J Clin Oncol. 2010;28:1547–53.
Kobayashi H, Tanisaka K, Doi O, Kodama K, Higashiyama M, Nakagawa H, et al. An in vitro chemosensitivity test for solid human tumors using collagen gel droplet embedded cultures. Int J Oncol 1997;11:449–455.
Kobayashi H. Development of a new in vitro chemosensitivity test using collagen gel droplet embedded culture and image analysis for clinical usefulness. Recent Results Cancer Res. 2003;161:48–61.
Koizumi W, Nakajima H, Hara T, Takagane A, Akiya T, Takagi M, et al. S-1 plus cisplatin versus S-1 alone for first-line treatment of advanced gastric cancer (SPIRITS trial): a phase III trial. Lancet Oncol. 2008;9:215–21.
Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomized controlled trial. Lancet. 2010;376:687–97.
Van Custum E, Köhne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29:2011–9.
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34.
Gu F, Ma Y, Zhang Z, Zhao J, Kobayashi H, Lun Z, Fu L. Expression of Stat3 and Notch1 is associated with cisplatin resistance in head and neck squamous cell carcinoma. Oncol Rep. 2010;23:671–6.
Higashiyama M, Oda K, Okami J, Maeda J, Kodanma K, Takami K, et al. In vitro-chemosensitivity test using the collagen gel droplet embedded culture drug test (CD-DST) for malignant pleural mesothelioma: possibility of clinical application. Ann Thorac Cardiovasc Surg 2008;14:355–362.
Higashiyama M, Oda K, Okami J, Maeda J, Kodama K, Imamura F, et al. Prediction of chemotherapeutic effect on postoperative recurrence by in vitro anticancer drug sensitivity testing in non-small lung cancer patients. Lung Cancer. 2010;68:472–7.
Kawamura M, Gika M, Abiko T, Inoue Y, Oyama T, Izumi Y, et al. Clinical evaluation of chemosensitivity test for patients with unresectable non-small cell lung cancer (NSCLC) using collagen gel droplet embedded culture drug sensitivity test (CD-DST). Cancer Chemother Pharmacol. 2007;59:507–13.
Higashiyama M, Kodama K, Yokouchi H, Takami K, Nakagawa H, Imamura F, et al. Cisplatin-based chemotherapy for postoperative recurrence in non-small cell lung cancer patient: relation of the in vitro chemosensitive test to clinical response. Oncol Rep. 2001;8:279–83.
Takamura Y, Kobayashi H, Taguchi T, Motoyama K, Inaji H, Noguchi S. Prediction of chemotherapeutic response by collagen gel droplet embedded culture-drug sensitivity test in human breast cancers. Int J Cancer. 2002;98:450–5.
Maejima K, Tokunaga A, Kiyama T, Kanno H, Bou H, Watanabe M, et al. Chemosensitivity test for 5-fluorouracil and 5-chloro-2,4-dihydroxypyridine predicts outcome of gastric cancer patients receiving S-1 postoperatively. Gastric Cancer. 2010;13:231–7.
Schrag D, Garewal HS, Burstein HJ, Samson DJ, Von Hoff DD, Somerfield MR. American Society of Clinical Oncology Technology Assessment: chemotherapy sensitivity and resistance assays. J Clin Oncol. 2004;22:3631–8.
Sakuramoto S, Sasako M, Yamaguchi T, Kinoshita T, Fujii M, Nashimoto A, et al. Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med. 2007;357:1810–20.
Kim KY, Koizumi K, Lee KH, Kishimoto T, Chung HC, Hara T, et al. Randomized phase III study of S-1 alone versus S-1 plus docetaxel (DOC) in the treatment for advanced gastric cancer (AGC): the START trial. J Clin Oncol. 2011;29 (abstr 7).
Narahara H, Iishi H, Tsuburaya A, Chin K, Imamoto H, Esaki T, et al. Randomized phase III study comparing the efficacy and safety of irinotecan plus S-1 with S-1 alone as first-line treatment for advanced gastric cancer (study GC0301/TOP-002). Gastric Cancer. 2011;14:72–80.
Wieand HS. Chemotherapy sensitivity and response assays: are the ASCO guidelines for clinical trial design too restrictive? J Clin Oncol. 2005;23:3643–4.
Acknowledgments
We thank the Future Medicine Promoting Network, a nonprofit organization established in Shiga Prefecture in 2004, for providing full financial support to this study. We thank the patients and their families and for participating in this study. Furthermore, we thank the following participating medical institutes and clinical staff: Ken Yanagihashi, Masato Kano (Otsu Municipal Hospital), Nobuo Baba (Japanese Red Cross Otsu Hospital), Toshikazu Yagi (Social Insurance Shiga Hospital), Masae Tsuji (Moriyama Municipal Hospital), Akira Kawaguchi (National Hospital Organization Shiga Hospital), Shoichi Nishimura (Yasu Hospital Association and the Medical Corporation), Masamitsu Hirano (Kusatsu general Hospital), Hisatomi Tanaka (Kohnan Hospital), Hideyuki Tsuzuki (Hikone Central Hospital), Yasushi Teramura (Hikone Municipal Hospital), Akira Yamamoto, Naoki Hayashi (Yujinkai Yujin-yamazaki Hospital), Ryuichi Irie (Takashima General Hospital), Yuji Kanda (Nagahama City Hospital), Tohru Yokota (Oumikusatsu Tokushukai Hospital), Kazuhiro Maruhashi, Takumi Shimomatsuya (Nagahama Red Cross Hospital), Masutsugu Fujita (Makino Hospital), Hitoshi Hasegawa (Biwako Ohashi Hospital), and Yoshihiro Tsutamoto (Toyosato Hospital).
Conflict of interest
None declared.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Naitoh, H., Yamamoto, H., Murata, S. et al. Stratified phase II trial to establish the usefulness of the collagen gel droplet embedded culture-drug sensitivity test (CD-DST) for advanced gastric cancer. Gastric Cancer 17, 630–637 (2014). https://doi.org/10.1007/s10120-013-0320-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-013-0320-4