
Abstract
Drug repurposing is a technique for probing new usages of existing medicines, but its traditional methods, such as computational approaches, can be time-consuming and laborious. Recently, knowledge graphs (KGs) have emerged as a powerful approach for graph-based representation in drug repurposing, encoding entities and relations to predict new connections and facilitate drug discovery. As COVID-19 has become a major public health concern, it is critical to establish an appropriate COVID-19 KG for drug repurposing to combat the spread of the virus. However, most publicly available COVID-19 KGs lack support for multi-relations and comprehensive entity types. Moreover, none of them originates from COVID-19-related drugs, making it challenging to identify effective treatments. To tackle these issues, we developed Drug-CoV, a drug-origin and multi-relational COVID-19 KG. We evaluated the quality of Drug-CoV by performing link prediction and comparing the results to another publicly available COVID-19 KG. Our results showed that Drug-CoV outperformed the comparing KG in predicting new links between entities. Overall, Drug-CoV represents a valuable resource for COVID-19 drug repurposing efforts and demonstrates the potential of KGs for facilitating drug discovery.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Drug repurposing is the process of discovering new uses for existing drugs that go beyond their original medical indication [3]. This approach is more efficient, cost-effective, and less risky than developing a completely new drug [70]. There are three steps in traditional drug repurposing methods: hypothesis generation, mechanistic assessment, and evaluation. Nonetheless, these methods are often time-consuming and cannot fully elucidate the underlying mechanisms between different biological entities, such as genes and proteins [40, 76].
The use of KGs in drug repurposing has recently been surging, owing to their ability to illuminate interconnections between entities and capture the semantics within different types of relationships [76]. KGs are a type of graph-structured knowledge base that encodes real-world entities and the relationships between them as triples (s, r, o), where s represents the subject, r represents the relation, and o represents the object. For example, (Nelfinavir, inhibitor, HIV) is a triple that encodes the relationship between the drug Nelfinavir and its inhibition of the HIV virus. KGs are capable of discovering new knowledge among entities with the help of knowledge graph embedding models (KGEs) [61]. KGEs learn to represent entities and relations in a low-dimensional space and then, perform a link prediction task, which involves inferring missing links between subject-object pairs [14]. This powerful approach enables efficient and effective drug repurposing by identifying potential new uses for existing drugs [78].
On the other hand, coronavirus disease 2019 (COVID-19) [73] has been listed as an international public health emergency by World Health Organization due to its high transmission rate [11]. Considering the daily increase in the number of COVID-19 patients, constructing an appropriate KG for COVID-19 drug repurposing has become urgent [17]. Previous studies [11, 17, 71, 76] have suggested that an appropriate KG should yield proper link prediction results on different KGEs. According to [27], embedding a KG with multi-relations can prove to be effective for extracting features of infrequent biological entities. Therefore, it is vital for a KG to contain multi-relations. Multi-relations refer to a set of relations (at least two) between an entity pair. For example, based on the introduction of IbuprofenFootnote 1 in the Comparative Toxicogenomics Database (CTD) [30], there are multi-relations of “marker/mechanism" and “therapeutic" between the drug Ibuprofen and the disease headache.
Many COVID-19 KGs are constructed to conduct drug repurposing for COVID-19. Some of them [12, 15, 62] extracted entities and relations from scientific papers. However, they have to filter unnecessary papers to improve the quality of the literature selection [62]. To avoid the literature selection problem, some works [1, 11, 17, 71] alternatively integrated multiple structured databases to form a new KG. We carried out a literature review on those papers and summarized their three limitations as follows. The details of the literature review are reported in Sect. 2.
Firstly, most publicly available COVID-19 KGs do not support multi-relations. Secondly, the entity and relation types in most existing COVID-19 KGs are inconsistent with those of general medical KGs, where entity types, such as drugs, diseases, genes, proteins, and side effects, are of interest [5, 70, 76]. Nevertheless, some existing COVID-19 KGs [1, 11, 17, 71] only include a subset of these entity types, which limits the KGE from discovering meaningful links. Additionally, we found that none of the existing COVID-19 KGs originated from COVID-19-related drugs, i.e., they integrated all COVID-19 information from existing databases, which might contain many irrelevant elements for drug repurposing. In general, drugs shown to be useful in previous trials are more important, i.e., building a KG originating from COVID-19 drugs can provide more valuable insights into COVID-19 drug repurposing [71].
In this article, we aimed to bridge the gap between the general medical domain and the COVID-19 domain by creating a drug-origin COVID-19 KG called Drug-CoV, for the COVID-19 drug repurposing task. Our KG can be found at https://github.com/SRL94/kg, and the construction method can also be applied to other drug-repurposing KGs. Our contributions are as follows:
-
1.
Establishing a dedicated COVID-19 KG, Drug-CoV, with multi-relations to evaluate biological relation prediction methods in COVID-19. By integrating data from four professional databases and extracting applicable triples, our Drug-CoV contains thousands of nodes containing drugs, diseases, genes, proteins, and side effects connected by a set of semantic relations.
-
2.
Measuring the quality of Drug-CoV by comparing the link prediction results between different KGEs. One external COVID-19 KG was also used to compare the quality of our KG and to evaluate KGEs systematically.
-
3.
Investigating the potential of Drug-CoV for drug repurposing by conducting drug repurposing with dropped triples. We ranked drug candidates for COVID-19 and analyzed these candidates from the biological perspective.
2 Literature review
In the literature review, we first introduce various artificial intelligence (AI)-based methods for drug repurposing. Then, we summarize the existing COVID-19 KGs and their limitations.
2.1 Artificial intelligence in drug repurposing
The era of big data has paved the way for using AI to define diseases, medicines and therapeutics, as well as to identify targets with high accuracy and precision [78]. This section summarizes the current AI-based drug repurposing methods, categorized into sequence-based and graph-based representations.
2.1.1 Sequence-based representation
Sequence-based representation methods [2, 21, 25, 69] use genetic or protein sequences as input data for machine learning models. These methods typically convert the sequences into numerical or vector representations, which can be processed by the machine learning model [39]. Machine learning utilizes hierarchical layers of linear and nonlinear transformations to investigate and analyze data [24, 28].
One example of a straightforward machine learning model is the artificial neural network (ANN), which draws inspiration from initial models of sensory processing in the brain [23]. ANN uses artificial neurons to nonlinearly transform the weighted sum of input feature variables to output targets. The weights are optimized by minimizing the prediction loss of the output targets through backpropagation on training samples [43]. For example, [2] employed ANN to classify drugs into therapeutic classes in pharmaceuticals by utilizing the drugs’ transcriptomic profile vectors. Lenselink et al. [25] compared the performance of various machine learning models in predicting targets and found that neural networks outperformed conventional machine learning methods like logistic regression. However, the generated vector representations of ANN are often high-dimensional and sparse [39].
To overcome the challenges posed by high-dimensional and sparse vector representations, more advanced models such as recurrent neural networks (RNNs) and convolutional neural networks (CNNs) have been developed to extract underlying features from biological sequences, resulting in improved performance [39]. For instance, [69] proposed a novel approach to drug repurposing that combines CNN and bidirectional long short-term Memory networks (LSTM). This approach uses the CNN module to learn the initial representation of drug-disease pairs based on their similarities and associations. Meanwhile, the LSTM module is employed to learn the path representations of the drug-disease pairs by using an attention mechanism [56] to balance the contributions of different paths. [21] developed DeepAffinity, a semi-supervised machine learning model that jointly encodes molecular representations using RNN and CNN.
2.1.2 Graph-based representation
Graph-based representation methods [44, 48] encode proteins and their chemical associations into a graph structure, where nodes represent molecules or atoms, and edges represent chemical bonds or interactions. These methods have gained popularity for link prediction in graphs, where nodes and edges are represented as low-dimensional vector representations. Using the vector representations of drugs and diseases, link prediction can measure their similarities and identify effective drugs for specific diseases [78]. Graph neural networks (GNNs) [77] and KGEs [61] are the two most popular techniques for learning vector representations of entities and relations in a KG.
GNNs extract latent features automatically by considering the neighboring nodes’ structure and aggregating information across multiple layers [77]. ProteinGCN [44] leverages the protein-ligand interaction graph to learn the features of the ligand and protein molecules and predict their binding affinity. To learn how drugs and diseases are embedded in different perspectives and predict new links between them, GCMM [75] constructs a graph by incorporating various known drug-disease relations, including drug-drug and disease-disease similarities, and uses a graph convolutional network (GCN) encoder. Nonetheless, GNNs may not perform well on graphs with sparse or irregular connectivity [64] since they heavily rely on the graph’s connectivity.
KGEs are utilized to capture global features, whereas GNNs mainly focus on preserving first-order or second-order proximity [39]. Mohamed et al. [35] proposed TriModel, a new KGE-based approach for predicting drug-target interactions in a multi-phase procedure, by extending DistMult [72] and ComplEx [53]. To predict links between diseases and drugs, [74] used the RotatE method proposed by [50] to acquire vector representations of entities (such as drugs and targets) and relationships (such as inhibition between drugs and targets).
In summary, graph-based representation methods have the advantage of capturing the complex relationships between drugs, targets, diseases, and other entities compared to sequence-based representation methods.
2.2 COVID-19 knowledge graphs
To enable drug repurposing for COVID-19 using graph-based representation, the construction of a suitable COVID-19 KG is crucial. We conducted a systematic review of existing COVID-19 KGs, using the search terms “COVID-19”, “knowledge graph”, and “drug repurposing”. We considered up to five papers returned by Google Scholar.
2.2.1 Knowledge graphs for information retrieval
There are two clusters of COVID-19 KGs. The first cluster includes KGs designed to facilitate information retrieval, such as those created by [9, 49, 62, 67]. These KGs capture the information in COVID-19-related scientific papers [58] and typically consist of paper nodes, author nodes, affiliation nodes, paper concept nodes, and more. They are used primarily for information retrieval and article recommendation [10]. For example, a typical query could be “Which papers discussing COVID-19 risk factors are most often cited by researchers within the CORD-19 dataset?” [67]. To ensure the quality of scientific papers, most KGs [9, 31, 49, 67] are constructed based on the CORD-19 dataset [60], which is a growing resource of scientific papers related to COVID-19 and historical coronavirus research.
2.2.2 Knowledge graphs for drug repurposing
The second cluster [1, 11, 17, 71] is designed to facilitate drug repurposing, which is one of the most critical tasks in the medical field as it can significantly speed up the traditional process of drug discovery and thereby shortens development timelines and reduces costs [22]. This cluster can be further split into two categories based on their sources.
The first category[12, 15, 62] extracts entities and relations from scientific papers. For instance, [15] created a cause-and-effect KG of COVID-19 pathophysiology to assist with drug repurposing. To create this KG, the literature was retrieved from open-access and freely available journals, such as PubMed and Europe PMC, with specific keywords. However, the literature selection quality remains a problem as the KGs have to filter literature based on available information about potential drug targets for COVID-19 [62].
The second category [1, 11, 17, 71] constructs KGs by integrating multiple existing KGs. For example, [71] integrated fourteen biological KGs. [11] added COVID-19-related information to an existing drug KG, rather than constructing a new one from scratch. [1] identified small molecules as potential drugs that target host proteins and disease processes involved in COVID-19. However, this requires experts to identify vocabulary, and the KG may contain irrelevant COVID-19 elements. Ge et al. [17] also constructed a public virus-related KG, called CoV-DTI, by integrating four public sources, but this KG only contains one relation, “interact with.”
In conclusion, our analysis reveals that there are some issues with the availability of KGs in the second category. Three out of the four reported KGs are not publicly available. Moreover, the publicly available KG lacks multi-relational information. Additionally, the entity types used in most COVID-19 KGs are inconsistent with those in general medical KGs. Furthermore, the KGs have integrated all COVID-19 information without filtering out irrelevant elements for drug repurposing. A KG specifically designed to identify useful COVID-19 drugs could provide more valuable insights for COVID-19 drug repurposing [41].
3 Concepts and notations
We now define the salient concepts that underlie Drug-CoV.
Knowledge graph. A KG is a type of structured data that represents knowledge as a graph. In this graph, nodes represent entities, and edges represent relationships or connections between them. A KG can be considered a type of semantic network that is used to organize and represent knowledge in a machine-readable format [66]. It represents information in the format of triples (subject, relation, object). Notable examples include Wikidata [59] and DrugBank [68]. The notation of the KG in this paper is denoted as \(G = (E, G)\), where E is the set of entities (e.g., drugs, diseases and genes) and R is the set of relations (e.g., cause, encode and target) that connect the entities.
Entity and relation. In a KG, entities refer to the real-world objects, concepts, or events that are being represented. Relations, on the other hand, describe the connections or interactions between entities in the real world. In a triple (subject, relation, object), the term subject (or object) also can be used interchangeably with the subject entity (or object entity). In this paper, a subject is denoted as \(s \in E\), an object is denoted as \(o \in E\) and a relation is denoted as \(r \in R\).
Multi-relation. In our paper, we adopt the definition of multi-relations introduced by [27]. Multi-relations refer to a situation where multiple types of relations (or edges) exist between a pair of entities.
Knowledge graph embedding. It is a technique to encode the entities and relations in a KG as dense and low-dimensional vector representations [4]. These vector representations, also called embeddings, capture the semantic meaning of the entities and relations in the graph, allowing for various downstream tasks such as link prediction. In this paper, we use the notation \(\mathbf {v_x}\) to represent a vector representation of item x.
Link prediction. Link prediction is the task of predicting the likelihood or probability of the existence of a relationship between two entities in a KG. KGE-generated embeddings can be used for link prediction, which has various applications, including predicting drug-target interactions in drug discovery [34].
More specifically, given a relation and an entity, the goal is to predict a missing entity, i.e., inferring s given (r, o) or inferring o given (s, r). To achieve this, KGEs calculate the score for each triple \((s, r, o_i)\) where \(o_i\) is a candidate entity from the entity set and then, select the entity with the highest score as the prediction result.
Attention mechanism. Attention mechanism is a concept used in neural networks that helps to focus on certain parts of the input while processing it [56]. It allows the network to assign different weights to different parts of the input, thus emphasizing or “paying attention" to more relevant information.
4 Methodology
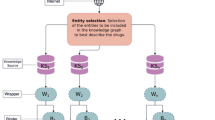
In this section, we first present an overview of our Drug-CoV construction and then, briefly introduce our multi-relational KGE, ConvMR. Figure 1 illustrates the five critical stages of Drug-CoV construction. Starting off, the drug seeding stage and database integration stage are gone through to build a skeleton KG by integrating structured information from multiple databases. The sentence extraction stage and triple extraction stage are then used to extract additional triples from documents. Finally, the combination stage merges the skeleton KG with the extracted triples. The resulting KG is stored in a graph database, Neo4j,Footnote 2 which stores nodes and edges rather than tables or documents.
4.1 Drug seeding
Yan et al. [71] demonstrated that useful drugs in prior trials are crucial for drug repurposing. To identify potential COVID-19 treatments, we retrieved information from DrugBank, a reliable and comprehensive web resource that provides detailed information about Food and Drug Administration (FDA)-approved drugs and experimental drugs in the FDA approval process.Footnote 3 We selected 68 drugs with COVID-19-related properties as seed entities for further analysis to extract other entities of interest. These drugs were chosen based on their prior use in treating diseases with similar symptoms to COVID-19 and their known mechanisms of action that could potentially be effective against the virus.

The construction overview of Drug-CoV
4.2 Database integration
In this stage, we aimed to identify additional entities of interest, such as genes, proteins, and diseases, as well as relevant relations among them that could be valuable for COVID-19 treatment.
Database integration involves two main steps: (1) entity extraction from multiple public databases and (2) entity resolution to identify and unify mentions of the same entity across different sources.
We extracted entities of interest from four public databases, namely DrugBank, PubChem [63], CTD, and MedlinePlus [32], which were chosen for their provision of high-quality structured information and their popularity in prior studies. These databases provided raw data files in various formats, including CSV, TSV and XML. To extract structured interactions between entity pairs (such as drug-disease and drug-drug interactions), we designed Python parsers that parse the raw data files based on their specific data structures.
To perform entity resolution, which involves identifying all mentions of the same entity across multiple knowledge bases [18], we unified drug names with chemical formulas (such as \({\textrm{C}}_{37}{\textrm{H}}_{48}{\textrm{N}}_{6}{\textrm{O}}_{5}{\textrm{S}}_{2}\) for the drug “Ritonavir”), gene names with symbols and NCBI Gene IDs, and protein names with Uniprot IDs. Disease and side effect names were extracted from DrugBank and MedlinePlus, respectively, due to their lack of standardization. Entity resolution prevented the duplication of entities in our KG resulting from synonyms in different resources.

The schema of our KG, Drug-CoV. Nodes are entity types. Edges are relation types between them
It is important to note that relations among entities were introduced from previous medical KGs [11, 70] to ensure the consistency of our COVID-19 KG with the general domain. Figure 2 shows the schema of our KG.
As a result, a KG is constructed by integrating structured data from multiple public databases. We will expand the KG in the following stages by extracting more triples from sentences.
4.3 Sentence extraction
In addition to structured data, the four databases also include detailed text descriptions. To extract relevant information, we focused on identifying sentences that mention at least two distinct entities. To achieve this, we developed a Python-based sentence extractor using Beautiful soupFootnote 4 and CoreNLP [29]. Beautiful soup helped us to parse the HTML/XML documents and extract relevant information while removing images, headers, and other elements from web pages. CoreNLP was utilized for both coreference resolution and sentence splitting. Coreference resolution is a technique adopted to identify linguistic expressions that refer to the same entity, which is essential for precise identification of sentences with interest.

An example of sentence splitting and coreference resolution in CoreNLP. CoreNLP automatically segments the input text into separate sentences and identifies all references to the same entity using consistent color labeling. In the second sentence, the pronoun “it" is replaced with “Ritonavir” for clarity. However, since the second sentence only contains one entity of interest, it would not be passed to the triple extraction stage
4.4 Triple extraction and combination
To extract triples from the sentences, we used two tools: BERN2 [51] and CoreNLP. BERN2 is a named entity recognition tool used in biomedical natural language processing that can recognize and normalize nine types of biomedical entities. Figure 4 shows an example of BERN2 use.

An example of biomedical named entity recognition in BERN2
On the other hand, CoreNLP was implemented to identify the relation between a pair of biomedical entities. Nonetheless, it is worth noting that the relations identified by CoreNLP might be out of the scope of our KG since it finds direct relations in the text. For instance, our model extracted (Dexamethasone, may cause, upset stomach) and (GC-373, therapeutic potential for, COVID-19) but “may cause" and “therapeutic potential for" are not included in the schema. Therefore, we designed a similarity-based method to normalize similar relations and filter out irrelevant ones.
Given a triple \((s_j, r_j, o_j)\) extracted by BERN2 and CoreNLP (e.g., (Dexamethasone, may cause, upset stomach)), we used Sentence-bert (SBERT) [42] to generate a vector representation for \(r_j\). Similarly, we generated vector representations for the ten relations in the schema (as shown in Fig. 2). With these vector representations, we can compare the cosine similarity between the relation \(r_j\) and every relation \(r_i \in R\). Then, we replaced \(r_j\) with the relation \(r_k \in R\) which has the highest similarity. This process can be represented by the following equations:
where the cosine similarity is represented by \(\cos \). Based on this similarity-based method, the triple (Dexamethasone, may cause, upset stomach) was normalized to (Dexamethasone, cause, upset stomach) using the method described above. Also, (GC-373, therapeutic potential for, COVID-19) was normalized to (GC-373, therapeutic, COVID-19).
In the combination stage, we added the triples extracted from sentences to the skeleton KG. Finally, we constructed Drug-CoV, which contains a total of 36,438 relations belonging to 10 different types, between 9694 entities of 5 different types.

The distributions of entity types in Drug-CoV
Figure 5 shows the percentage of each entity type. The “Gene” category comprised 52%, the largest percentage of total entity types since it can indicate many potential paths between two different diseases. The “Drug" category made up 35%, the second largest percentage of total entity types since Drug-CoV is a drug-origin-based KG and can provide more insights between two different drugs.
4.5 Convolutional and multi-relational knowledge graph embedding
We proposed a Convolutional and Multi-relational model (ConvMR) [27] that enhances the semantic connection between multi-relations. We briefly introduce the encoding and convolution part as follows.
In learning, ConvMR concerns not only direct connections between an entity pair (i.e., original triples of a KG: \((s, r_1, o), (s, r_2, o),\ldots , (s, r_N, o)\)) but also multi-relations between the pair (i.e., \((s, r_1, r_2,\ldots , r_N, o)\)). To clarify, we call the original triple (s, r, o) as triple; triple with multi-relation \((s, r_1, r_2,\ldots , r_N, o)\) as multi-relation triple, which means there are a total of N relations between (s, o) and \(N \ge 2\). Given a triple or a multi-relation triple, ConvMR encodes relations by using a proposed attention-based average operation (attn-average). Generally, the attn-average uses an attention mechanism to assign weights to relations among an entity pair and then, encodes them into a relation vector \(\mathbf {v_{r'}} \in \mathbb {R}^{1 \times M}\), where M is the dimension of the vector.
In the convolution part, ConvMR employs a convolution layer to calculate the score for the multi-relation triple \((s, r', o)\):
where concat is the concatenation operation. \(\Omega \) is a set of filters. Filters in convolutional layers are small matrices or tensors that are applied to an input matrix in a sliding window fashion. Each filter learns to detect specific features or patterns in the input. \(*\) is the convolution operation; g is the activation function ReLU. In training, valid triples or multi-relation triples are assigned higher scores than invalid triples or multi-relation triples.
In summary, the proposed ConvMR addresses the connection of multi-relations between an entity pair by learning their weighted joint. Compared to traditional KGEs, ConvMR can maintain the semantic connection between multi-relations and effectively mine features of less frequent entities.
5 Experiments
In this section, we measure the quality of Drug-CoV by comparing link prediction results between different KGEs and investigate the capacity of the Drug-CoV for drug repurposing.
5.1 KGE baselines
We used LibKGE [6], an open-source Python package for KGE, including TranE [4], RESCAL [38], TransH [65], DistMult [72], ComplEx [53], ConvE [14] and RotatE [50] in the comparison study. We chose these KGEs because they are the most popular KGEs in previous drug repurposing works [16, 37, 74, 76] and they can represent different kinds of KGEs, such as distance-based models (TransE, TransH), neural network models (ConvE) and semantic matching models (RESCAL, DistMult, ComplEx and RotatE). In order to handle the multi-relations, we used ConvMR, which can extract the semantic hierarchical information in multi-relations to enhance KGE. We also implemented a GNN methods, CompGCN [55], as well as several recent neural network-based and semantic matching-based methods, including GIE [8], MuRT [13] and HousE [26].
5.2 Dataset
In order to provide a fair comparison of these baselines, we included CoV-DTI [17]Footnote 5 to perform further evaluations. CoV-DTI is a virus-based KG that is constructed by integrating different databases. As illustrated in Sect. 2, the availability of the existing COVID-19 KGs is a problem, and CoV-DTI is the only public COVID-19 KG targeting drug repurposing with a similar size. CoV-DTI does not contain multi-relations. The statistical information of this dataset is shown in Table 1.
5.3 Training protocols
We divided the triples into a training set, a validation set and a test set in an 8:1:1 manner. All the baselines from LibKGE were trained with the learning rate of [0.001, 0.01, 0.1] and the dimension of the initial embedding of [100, 200]. Other hyper-parameters for each approach were set at their default settings, as recommended by the LibKGE package. ConvMR, CompGCN, GIE, MuRT, and HousE were trained with the learning rate [0.001, 0.01, 0.1], the dimension of the initial embedding of [100, 200], and the epochs of [20, 30]. We implemented the grid search for parameter optimization.
5.4 Evaluation metrics
For the quality evaluation of Drug-CoV, we performed the link prediction task, as suggested by the KG community [4, 50]. For each test triple, the s or o is replaced by every other entity \(e \in E\). We first computed a similarity score for each test triple and then, sorted these scores in descending order to get the rank of the correct test triple. We used mean reciprocal rank (MRR) and the proportion of correct entities in the top N ranks (Hits@N) for \(N =1, 10\) and 100. Higher MRR and Hits@N values are indications of a better performance. Note that we used the filtered setting protocol [4] in our experiments, i.e., not taking any corrupted triples into account. The “corrupted triple" means the triple appears in the KG.
To evaluate Drug-CoV’s capacity for drug repurposing, we performed link prediction on a specific relation, “therapeutic".
5.5 Evaluation of KGs with different embedding methods
We tested different embedding methods on Drug-CoV and CoV-DTI. As shown in Table 2, ConvMR outperformed all other models regarding Hits@N and MRR on Drug-CoV. Generally, compared with other baselines, ConvMR can improve the link prediction performance by maintaining the semantic hierarchy of multi-relations [27]. For example, ComplEx achieved a 1.6 (49.4\(-\)47.8)% improvement in terms of Hits@100 value compared with TransE. TransH obtained a 8.2 (46.4\(-\)38.2)% increment in the Hits@100 when compared with RESCAL. Considering the multi-relation features, ConvMR exceeded the ComplEx by 3.3 (25.3\(-\)22.0)% (MRR), 2.8 (17.5\(-\)14.7)% (Hits@1), 4.3 (40.7\(-\)36.4)% (Hits@10) and 13.9 (63.3\(-\)49.4)% (Hits@100). It also surpassed TransH with 18.2 (25.3\(-\)7.1)% (MRR), 15.4 (17.5\(-\)2.1)% (Hits@1), 23.8 (40.7\(-\)16.9)% (Hits@10) and 16.9 (63.3\(-\)46.4)% (Hits@100). These results demonstrate that ConvMR is more effective and could be used on biological relation prediction tasks to improve prediction performance.
A similar trend can be seen in evaluating CoV-DTI; however, ConvMR did not achieve the best performance on all metrics. It is mainly because CoV-DTI does not have multi-relation features for ConvMR to encode. Compared with other neural network-based models, such as ConvE, ConvMR still performed better on all metrics, especially for Hits@100 (57.2\(-\)12.1 = 45.1% improvement).
We also found that the best performance of CoV-DTI and Drug-CoV was not significantly different. Compared with CoV-DTI, Drug-CoV contains more complex relation types and entity types that can form more relational paths between entities. This result demonstrates that Drug-CoV facilitates the modeling of COVID-19 terms.
5.6 Ablation study of multi-relations
In this section, we study the importance of encoding multi-relation features in Drug-CoV. ConvMR is the only model capable of encoding multi-relations among all the baselines. ConvMR uses an average-based attention mechanism to assign different weights to reveal the semantic/hierarchical connection between multi-relations. We conducted an ablation study by omitting the attention mechanism. The ablated model (ConvMR-w/o) was found to cause a drop in the results by decreasing all metrics 1.8 (25.3\(-\)23.5)% MRR, 2.1 (17.5\(-\)15.4)% Hits@1, 0.2 (40.7\(-\)40.5)% Hits@100, 2.2 (63.3\(-\)61.1)% Hits@100. These decreases suggest that encoding the connection between multi-relations plays a pivotal role in relation prediction in the medical domain.
We further randomly selected one multi-relation triple (Ibuprofen, marker/mechanism, therapeutic, Headache) from Drug-CoV and fed it into the trained attention mechanism. The attention mechanism assigned 0.85 weight to relation “marker/mechanism" and 0.15 to relation “therapeutic”. According to the referencs on the database CTD,Footnote 6 we found that 2 out of 3 references labelled “Ibuprofen” as a “marker/mechanism” to “Headache”, and 1 out of 3 labelled “Ibuprofen” as a “treatment” to “Headache”. Based on this observation, we can conclude that the weights reflect the importance of a relation between an entity pair in the medical domain.
5.7 Evaluation of Drug-CoV’s capacity for drug repurposing
Drug repurposing is one of the most widely used application in the biological field. Previous studies [17, 76] did not specify how they dropped drug-disease links and what the drop rate was. In our work, we randomly dropped 10% drug-disease links with relation “therapeutic" from the original KG. The dropped 10% links were testing data and the original KG without the 10% link were used to train models.
As shown in Table 4, ConvMR achieved a Hits@100 of 96.6%, significantly outperforming that of RESCAL (Hits@100 = 31.0%), DistMult (Hits@100 = 34.5%) and ComplEx (Hits@100 = 94.8%). If compared with MRR, Hits@1 and Hits@10, ConvMR also attained a top-five performance. For example, concerning MRR, the ranked top-five models are TransE (47.2%), ConvE (47.0%), ComplEx (39.2%), ConvMR (31.0%) and CompGCN (30.9%). As for Hits@10, the ranked top-five models are ConvE (86.9%), ComplEx (84.5%), ConvMR (82.8%), TransE (82.8%) and GIE (50.0%). The superior performance of ConvMR likely results from the full use of semantic information available in multi-relations. TransE outperformed other models on MRR and Hits@1, indicating many one-to-one relations [65] in Drug-CoV after dropping drug-disease links. The large amount of one-to-one relations also restricted the ability of ConvMR to mine information hidden behind multi-relations. The proper results of ConvE and ConvMR mean that convolution-based models are better at extracting local information in the medical domain.
In summary, most models performed well in distinguishing drug-disease links in the drug repurposing task. Therefore, Drug-CoV is useful for modelling COVID-19 items and providing new insights into drug repurposing predictions.
5.8 Extraction and ranking of drug candidates for COVID-19
To test the COVID-19 drug repurposing ability of our KG Drug-CoV, we employed ConvMR and embedding scores to rank candidate drugs. We then ranked the top ten drug candidates for COVID-19 repurposing based on embedding scores. The embedding score of drug x could be calculated by the equation below:
where CovMR(.) is the prediction score of the triple. The prediction score is in the range between 0 and 1. A higher score represents a stronger potential association between the drug and COVID-19.
The resulting top ten predicted drug candidates are listed in Table 5. Anti-inflammatory drugs are the most commonly suggested candidates among the top ten. For example, Baricitinib is a drug that inhibits Janus kinases (JAKs), which are enzymes involved in inflammation and immune responses [33]. JAK inhibition was identified as a treatment strategy for COVID-19 [45,46,47]. A study by [52] analyzed eight cohort studies and five randomized controlled trials of Baricitinib compared to control groups, looking at clinical and laboratory parameters. The meta-analysis results showed a significant reduction in mortality, improved \(PaO_2/FiO_2\) ratio, and lower C-reactive protein (CRP) levels in the Baricitinib group compared to the control group. Based on these findings, the study suggests using Baricitinib for moderate to severe COVID-19 cases.
Angiotensin-converting enzyme 2 (ACE2) is an enzyme that plays a role in regulating blood pressure [19]. ACE2 has been identified as a receptor for SARS-CoV-2, the virus that causes COVID-19, allowing the virus to enter host cells [57]. Ibuprofen, a non-steroidal anti-inflammatory agent, can increase ACE2 expression in rat and culture models [54]. However, the implications of this in relation to COVID-19 are still uncertain and further studies are needed to determine if this impacts the course of the disease. Additionally, the use of Ibuprofen in COVID-19 patients has been controversial, with some studies suggesting that it may increase the risk of severe illness or death in certain populations [36].
Our results highlight the potential of several anti-infective drugs, including Lopinavir, Ritonavir, and Darunavir. These drugs can target the 3C-like protease (3CLpro), a major protease in CoV that cleaves the large replicase polyproteins during viral replication. Lopinavir and Ritonavir are protease inhibitors that can effectively target 3CLpro [20]. However, clinical trials of these drugs have yielded mixed results. Some studies have shown no benefit in terms of reducing mortality or time to clinical improvement, while others have shown a modest benefit in hospitalized patients [7].
Our results on drug repurposing generate hypotheses regarding which existing drugs have greater potential for repurposing in treating COVID-19. Although the KG does not provide clinical or biological evidence, it provides valuable insights.
6 Conclusion
In this work, a dedicated COVID-19 KG called Drug-CoV was constructed for drug repurposing. By integrating and extracting information from multiple public databases, we have constructed a KG with over 30,000 interconnections between drugs, diseases, side effects, genes, and proteins. Our work is innovative in that it is the first to provide insights into multi-relations in drug repurposing. We have also demonstrated the effectiveness of encoding multi-relations in improving link prediction task performance. Our KG considers the importance of useful drugs and constructs a drug-origin KG. The proper results of experiments indicate that Drug-CoV can facilitate the modelling of COVID-19 terms on biological relation prediction and the repurposing of potential drugs.
Moreover, our KG can be expanded with more triples for future COVID-19 studies, thereby facilitating other research in this area. In addition, our KG construction methodology can be applied to discover effective treatments for other viruses, and it can assist in identifying the most promising drug candidates for further investigation.
Notes
Available from https://go.drugbank.com/COVID-19.
Available from https://github.com/FangpingWan/CoV-DTI.
References
Al-Saleem J, Granet R, Ramakrishnan S et al (2021) Knowledge graph-based approaches to drug repurposing for covid-19. J Chem Inf Model 61(8):4058–4067
Aliper A, Plis S, Artemov A et al (2016) Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data. Mol Pharm 13(7):2524–2530
Ashburn TT, Thor KB (2004) Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov 3(8):673–683
Bordes A, Usunier N, Garcia-Duran A, et al (2013) Translating embeddings for modeling multi-relational data. In: Advances in neural information processing systems, vol 26
Boudin M (2020) Computational approaches for drug repositioning: towards a holistic perspective based on knowledge graphs. In: Proceedings of the 29th ACM international conference on information and knowledge management, pp 3225–3228
Broscheit S, Ruffinelli D, Kochsiek A, et al (2020) LibKGE - A knowledge graph embedding library for reproducible research. In: Proceedings of the 2020 conference on empirical methods in natural language processing: system demonstrations, pp 165–174. https://www.aclweb.org/anthology/2020.emnlp-demos.22
Cao B, Wang Y, Wen D, et al (2020) A trial of lopinavir–ritonavir in adults hospitalized with severe covid-19. New England J Med
Cao Z, Xu Q, Yang Z, et al (2022) Geometry interaction knowledge graph embeddings. In: Thirty-sixth AAAI conference on artificial intelligence, AAAI 2022, thirty-fourth conference on innovative applications of artificial intelligence, IAAI 2022, the twelveth symposium on educational advances in artificial intelligence, EAAI 2022 Virtual Event, February 22–March 1, 2022. AAAI Press, pp 5521–5529. https://ojs.aaai.org/index.php/AAAI/article/view/20491
Cernile G, Heritage T, Sebire NJ et al (2021) Network graph representation of covid-19 scientific publications to aid knowledge discovery. BMJ Health Care Inf 28:1
Chatterjee A, Nardi C, Oberije C et al (2021) Knowledge graphs for covid-19: an exploratory review of the current landscape. J Personal Med 11(4):300
Che M, Yao K, Che C et al (2021) Knowledge-graph-based drug repositioning against covid-19 by graph convolutional network with attention mechanism. Future Internet 13(1):13
Chen C, Ross KE, Gavali S et al (2021) Covid-19 knowledge graph from semantic integration of biomedical literature and databases. Bioinformatics 37(23):4597–4598
Choudhary N, Reddy CK (2022) Towards scalable hyperbolic neural networks using Taylor series approximations. CoRR arXiv:2206.03610
Dettmers T, Minervini P, Stenetorp P, et al (2018) Convolutional 2d knowledge graph embeddings. In: McIlraith SA, Weinberger KQ (eds) Proceedings of the Thirty-Second AAAI conference on artificial intelligence, (AAAI-18), the 30th innovative applications of artificial intelligence (IAAI-18), and the 8th AAAI symposium on educational advances in artificial intelligence (EAAI-18), New Orleans, Louisiana, USA, February 2–7, 2018. AAAI Press, pp 1811–1818. https://www.aaai.org/ocs/index.php/AAAI/AAAI18/paper/view/17366
Domingo-Fernández D, Baksi S, Schultz B et al (2021) Covid-19 knowledge graph: a computable, multi-modal, cause-and-effect knowledge model of covid-19 pathophysiology. Bioinformatics 37(9):1332–1334
Gao Z, Ding P, Xu R (2022) Kg-predict: a knowledge graph computational framework for drug repurposing. J Biomed Inform 132(104):133
Ge Y, Tian T, Huang S et al (2021) An integrative drug repositioning framework discovered a potential therapeutic agent targeting covid-19. Signal Transduct Target Ther 6(1):1–16
Getoor L, Machanavajjhala A (2013) Entity resolution for big data. In: Proceedings of the 19th ACM SIGKDD international conference on knowledge discovery and data mining, pp 1527–1527
Hamming I, Cooper ME, Haagmans BL et al (2007) The emerging role of ace2 in physiology and disease. J Pathol J Pathol Soc Great Britain Ireland 212(1):1–11
Horby PW, Mafham M, Bell JL et al (2020) Lopinavir-ritonavir in patients admitted to hospital with covid-19 (recovery): a randomised, controlled, open-label, platform trial. The Lancet 396(10259):1345–1352
Karimi M, Wu D, Wang Z et al (2019) Deepaffinity: interpretable deep learning of compound-protein affinity through unified recurrent and convolutional neural networks. Bioinformatics 35(18):3329–3338
Kejriwal M (2020) Knowledge graphs and covid-19: opportunities, challenges, and implementation. Harv Data Sci Rev 11:300
Krogh A (2008) What are artificial neural networks? Nat Biotechnol 26(2):195–197
LeCun Y, Bengio Y, Hinton G (2015) Deep learning. Nature 521(7553):436–444
Lenselink EB, Ten Dijke N, Bongers B et al (2017) Beyond the hype: deep neural networks outperform established methods using a chembl bioactivity benchmark set. J Cheminf 9(1):1–14
Li R, Zhao J, Li C, et al (2022a) House: knowledge graph embedding with householder parameterization. In: Chaudhuri K, Jegelka S, Song L, et al (eds) International conference on machine learning, ICML 2022, 17–23 July 2022, Baltimore, Maryland, USA, Proceedings of machine learning research, vol 162. PMLR, pp 13209–13224. https://proceedings.mlr.press/v162/li22ab.html
Li S, Wong KW, Zhu D, et al (2022b) Modelling multi-relations for convolutional-based knowledge graph embedding. Procedia Comput Sci 207:624–633. https://doi.org/10.1016/j.procs.2022.09.117, https://www.sciencedirect.com/science/article/pii/S187705092200998X, knowledge-based and intelligent information and engineering systems: proceedings of the 26th international conference KES2022
Liu J, Huang J, Zhou Y et al (2022) From distributed machine learning to federated learning: a survey. Knowl Inf Syst 64(4):885–917
Manning CD, Surdeanu M, Bauer J, et al (2014) The stanford corenlp natural language processing toolkit. In: Proceedings of 52nd annual meeting of the association for computational linguistics: system demonstrations, pp 55–60
Mattingly CJ, Colby GT, Forrest JN et al (2003) The comparative toxicogenomics database (ctd). Environ Health Perspect 111(6):793–795
Michel F, Gandon F, Ah-Kane V, et al (2020) Covid-on-the-web: Knowledge graph and services to advance covid-19 research. In: International semantic web conference. Springer, pp 294–310
Miller N, Lacroix EM, Backus JE (2000) Medlineplus: building and maintaining the national library of medicine’s consumer health web service. Bull Med Libr Assoc 88(1):11
Mogul A, Corsi K, McAuliffe L (2019) Baricitinib: the second fda-approved jak inhibitor for the treatment of rheumatoid arthritis. Ann Pharmacother 53(9):947–953
Mohamed SK, Nounu A, Nováček V (2019) Drug target discovery using knowledge graph embeddings. In: Proceedings of the 34th ACM/SIGAPP symposium on applied computing, pp 11–18
Mohamed SK, Nováček V, Nounu A (2020) Discovering protein drug targets using knowledge graph embeddings. Bioinformatics 36(2):603–610
Moore N, Carleton B, Blin P, et al (2020) Does ibuprofen worsen covid-19?
Nian Y, Hu X, Zhang R et al (2022) Mining on Alzheimer’s diseases related knowledge graph to identity potential ad-related semantic triples for drug repurposing. BMC Bioinf 23(6):1–15
Nickel M, Tresp V, Kriegel HP (2011) A three-way model for collective learning on multi-relational data. In: Icml
Pan X, Lin X, Cao D et al (2022) Deep learning for drug repurposing: methods, databases, and applications. Wiley Interdiscip Rev Comput Mol Sci 12(4):e1597
Pushpakom S, Iorio F, Eyers PA et al (2019) Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov 18(1):41–58
Ratajczak F, Joblin M, Ringsquandl M et al (2022) Task-driven knowledge graph filtering improves prioritizing drugs for repurposing. BMC Bioinf 23(1):1–19
Reimers N, Gurevych I (2019) Sentence-bert: sentence embeddings using siamese bert-networks. In: Proceedings of the 2019 conference on empirical methods in natural language processing and the 9th international joint conference on natural language processing (EMNLP-IJCNLP), pp 3982–3992
Rumelhart DE, Hinton GE, Williams RJ (1986) Learning representations by back-propagating errors. Nature 323(6088):533–536
Sanyal S, Anishchenko I, Dagar A, et al (2020) Proteingcn: protein model quality assessment using graph convolutional networks. BioRxiv, pp 2020–04
Satarker S, Tom AA, Shaji RA et al (2021) Jak-stat pathway inhibition and their implications in covid-19 therapy. Postgrad Med 133(5):489–507
Seif F, Aazami H, Khoshmirsafa M et al (2020) Jak inhibition as a new treatment strategy for patients with covid-19. Int Arch Allergy Immunol 181(6):467–475
Seror R, Camus M, Salmon JH et al (2022) Do jak inhibitors affect immune response to covid-19 vaccination? data from the majik-sfr registry. Lancet Rheumatol 4(1):e8–e11
Sosa DN, Derry A, Guo M, et al (2019) A literature-based knowledge graph embedding method for identifying drug repurposing opportunities in rare diseases. In: Pacific symposium on biocomputing 2020, World Scientific, pp 463–474
Steenwinckel B, Vandewiele G, Rausch I, et al (2020) Facilitating the analysis of covid-19 literature through a knowledge graph. In: International semantic web conference. Springer, pp 344–357
Sun Z, Deng Z, Nie J, et al (2019) Rotate: knowledge graph embedding by relational rotation in complex space. In: 7th International conference on learning representations, ICLR 2019, New Orleans, LA, USA, May 6–9, 2019. OpenReview.net. https://openreview.net/forum?id=HkgEQnRqYQ
Sung M, Jeong M, Choi Y, et al (2022) Bern2: an advanced neural biomedical named entity recognition and normalization tool. arXiv:2201.02080
Tahsini Tekantapeh S, Ghojazadeh M, Ghamari AA et al (2022) Therapeutic and anti-inflammatory effects of baricitinib on mortality, icu transfer, clinical improvement, and crs-related laboratory parameters of hospitalized patients with moderate to severe covid-19 pneumonia: a systematic review and meta-analysis. Expert Rev Respir Med 16(10):1109–1132
Trouillon T, Welbl J, Riedel S, et al (2016) Complex embeddings for simple link prediction. In: International conference on machine learning, PMLR, pp 2071–2080
Valenzuela R, Pedrosa MA, Garrido-Gil P, et al (2021) Interactions between ibuprofen, ace2, renin-angiotensin system, and spike protein in the lung. implications for covid-19. Clin Transl Med 11(4)
Vashishth S, Sanyal S, Nitin V, et al (2020) Composition-based multi-relational graph convolutional networks. In: 8th International conference on learning representations, ICLR 2020, Addis Ababa, Ethiopia, April 26–30, 2020. OpenReview.net. https://openreview.net/forum?id=BylA_C4tPr
Vaswani A, Shazeer N, Parmar N, et al (2017) Attention is all you need. In: Advances in neural information processing systems, vol 30
Verdecchia P, Cavallini C, Spanevello A et al (2020) The pivotal link between ace2 deficiency and sars-cov-2 infection. Eur J Intern Med 76:14–20
Vergoulis T, Kanellos I, Chatzopoulos S et al (2021) Bip4covid19: releasing impact measures for articles relevant to covid-19. Quant Sci Stud 2(4):1447–1465
Vrandečić D, Krötzsch M (2014) Wikidata: a free collaborative knowledgebase. Commun ACM 57(10):78–85
Wang LL, Lo K, Chandrasekhar Y, et al (2020) Cord-19: the covid-19 open research dataset. ArXiv
Wang Q, Mao Z, Wang B et al (2017) Knowledge graph embedding: a survey of approaches and applications. IEEE Trans Knowl Data Eng 29(12):2724–2743
Wang Q, Li M, Wang X, et al (2021a) Covid-19 literature knowledge graph construction and drug repurposing report generation. In: Proceedings of the 2021 conference of the North American chapter of the association for computational linguistics: human language technologies: demonstrations, pp 66–77
Wang Y, Xiao J, Suzek TO et al (2009) Pubchem: a public information system for analyzing bioactivities of small molecules. Nucl Acids Res 37(suppl 2):W623–W633
Wang Y, Feng B, Ding Y (2021b) Tc-gnn: Accelerating sparse graph neural network computation via dense tensor core on gpus. arXiv:2112.02052
Wang Z, Zhang J, Feng J, et al (2014) Knowledge graph embedding by translating on hyperplanes. In: Proceedings of the AAAI conference on artificial intelligence
Weller K (2010) Knowledge representation in the social semantic web. In: Knowledge representation in the social semantic web. KG Saur
Wise C, Calvo MR, Bhatia P, et al (2020) Covid-19 knowledge graph: accelerating information retrieval and discovery for scientific literature. In: Proceedings of knowledgeable NLP: the first workshop on integrating structured knowledge and neural networks for NLP, pp 1–10
Wishart DS, Feunang YD, Guo AC et al (2018) Drugbank 5.0: a major update to the drugbank database for 2018. Nucl Acids Res 46(D1):D1074–D1082
Xuan P, Ye Y, Zhang T et al (2019) Convolutional neural network and bidirectional long short-term memory-based method for predicting drug-disease associations. Cells 8(7):705
Xue H, Li J, Xie H et al (2018) Review of drug repositioning approaches and resources. Int J Biol Sci 14(10):1232
Yan VK, Li X, Ye X et al (2021) Drug repurposing for the treatment of covid-19: a knowledge graph approach. Adv Ther 4(7):2100055
Yang B, Yih W, He X, et al (2015) Embedding entities and relations for learning and inference in knowledge bases. In: Bengio Y, LeCun Y (eds) 3rd International conference on learning representations, ICLR 2015, San Diego, CA, USA, May 7–9, 2015, conference track proceedings. http://arxiv.org/abs/1412.6575
Yang L, Liu S, Liu J et al (2020) Covid-19: immunopathogenesis and immunotherapeutics. Signal Transduct Target Ther 5(1):128
Zeng X, Song X, Ma T et al (2020) Repurpose open data to discover therapeutics for covid-19 using deep learning. J Proteome Res 19(11):4624–4636
Zhang F, Hu W, Liu Y (2022) Gcmm: graph convolution network based on multimodal attention mechanism for drug repurposing. BMC Bioinf 23(1):1–17
Zheng S, Rao J, Song Y et al (2021) Pharmkg: a dedicated knowledge graph benchmark for bomedical data mining. Brief Bioinform 22(4):344
Zhou J, Cui G, Hu S et al (2020) Graph neural networks: a review of methods and applications. AI Open 1:57–81
Zhou Y, Wang F, Tang J et al (2020) Artificial intelligence in covid-19 drug repurposing. Lancet Digital Health 2(12):e667–e676
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions
Author information
Authors and Affiliations
Contributions
SL wrote the main manuscript. KWW, DZ, CF supervised and guided this work. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, S., Wong, K.W., Zhu, D. et al. Drug-CoV: a drug-origin knowledge graph discovering drug repurposing targeting COVID-19. Knowl Inf Syst 65, 5289–5308 (2023). https://doi.org/10.1007/s10115-023-01923-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10115-023-01923-5