
Abstract
Obesity and overweight is a global health crisis and novel methods of treatment are needed to address it. Low-level laser therapy (LLLT) is a currently available non-invasive procedure for lysing excess fat, but there is a lack of consensus exists on LLLT frequency and limited research from studies of LLLT. The purpose of this pilot study is to compare the effect of three of the most common LLLT frequencies on weight, waist circumference, body fat percentage, and quality of life. Sixty overweight (body mass index (BMI) 25–29.9 kg/m2) adult participants were randomized to 12 LLLT treatments: (1) three times weekly for 4 weeks, (2) twice weekly for 6 weeks, or (3) once weekly for 12 weeks. All participants attended an in-person visit at baseline and at weeks 4, 6, 12, and 26. Participants were recruited September 30, 2016 through to August 27, 2017. The majority of the 60 participants were female (90%) with an average age of 43.7 years (± 9.2 years). Most participants (98%) completed 10 or more of the 12 LLLT treatments. When comparing across treatment groups, the greatest reductions from baseline were observed in those assigned to twice weekly for 6 weeks in weight (1 ± 1.7 (±SD) kg by week 6), waist circumference (− 2.0 ± 3.2 in. by week 6 and − 1.5 ± 3.2 in. by week 26), body mass index (− 0.4 ± 0.6 kg/m2), and body fat mass (− 1.1 ± 1.6 kg). This group also had the most significant improvement from baseline in quality of life (+ 0.5 ± 0.8 by week 6), body satisfaction (+ 0.2 ± 0.4 by week 6 and week 26), and body appreciation (+ 0.2 ± 0.3 by week 6 and + 0.3 ± 0.3 by week 26). LLLT twice weekly for 6 weeks could be proposed as the optimal frequency and duration for the management of body weight. Trial registration: https://clinicaltrials.gov/ct2/show/NCT02877004. Registered August 24, 2016.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Currently in the USA, about 97 million adults are overweight, accounting for about 33% of the adult population [1]. Low-level laser therapy (LLLT) is a novel non-invasive procedure that lyses excess fat without the negative side effects associated with surgical methods such as liposuction. LLLT has been associated with reductions in waist circumference of 6–12 in. with six treatments. LLLT has the potential to enhance motivation for weight management treatments by providing immediate positive feedback through reductions in waist circumference recognizable by the individual.
A variety of mechanisms of action have been proposed to explain how LLLT reduces body fat [2]. The two most popular theories include the lysing of the adipocytes producing transient pores in the cell membrane allowing lipids to flow out [3] and the initiation of lipid peroxidation by LLLT, which temporarily damages the cellular membrane of the adipocytes by creating pores [4]. Other potential mechanisms are discussed in recent review by Avci et al. [2]
Clinically, LLLT treatments can vary from 6 to 28 treatments with a frequency of 1 to 3 times per week depending on the participant’s preference and ability to cover the costs of the LLLT and the LLLT device used. In an earlier study, our research team conducted a feasibility study of 45 participants who are overweight or obese evaluating the use of 12 LLLT treatments over the course of 12 weeks (once per week) with the goal of reducing central adiposity [5]. The LLLT device used 532-nm green diodes. LLLT was compared with lorcaserin alone or in combination with LLLT. Participants were randomized to one of three conditions: LLLT, lorcaserin, and LLLT + lorcaserin. Although the sample size was too small for any statistically significant findings, the LLLT was observed to be associated with a reduction in body circumference (2.3–4.0 cm reduction) and a reduction in weight (1–3.5 kg reduction) with no identified significant adverse events. Another earlier study by Jackson et al., which used the same LLLT device (532-nm green diodes) on 68 subjects and randomized them to LLLT or sham treatment 3 times a week for 2 weeks, showed that the LLLT subjects had a greater decrease in body weight (p < 0.0005) and BMI (p < 0.001) [6].
Other research has been undertaken an earlier LLLT device (635-nm red diodes instead of 532-nm green diodes). In the same year, McRae et al. corroborated these findings with 86 patients retrospectively treated with LLT for over 2 weeks. Each patient received six treatments evenly spaced over 2 weeks (20 min in front and 20 min in back). In this study, they found a mean reduction of 1.24 lb (p < 0.0001) [7]. A year prior, Jackson had conducted a study of 689 subjects using LLLT 3 times a week for 2 weeks (six treatments) and reported significant reduction in circumference of waist, hips, and thighs on average by 5.17 in. between baseline and post treatment (p < 0.0001) [8]. These significant reductions in circumference were also noted by Nestor et al. in 80 patients in the same year over six treatments, but no change in BMI was observed [9].
Uncertainty remains regarding the ideal frequency of LLLT treatments needed for a significant weight loss and body circumference reduction. The purpose of this current study was to obtain preliminary results regarding the most efficacious frequency of LLLT treatments to produce body circumference reduction and weight loss.
Methods
Study overview
The present study was an open-label clinical trial in which participants received 12 LLLT treatments. To keep the number of treatments consistent across study arms, study participants were randomized to one of three treatment frequencies: (1) three times a week for 4 weeks (group A); (2) twice a week for 6 weeks (group B); (3) once a week for 12 weeks (group C). This study was approved by the Mayo Clinic Institutional Review Board and written informed consent was obtained for all study participants.
Setting
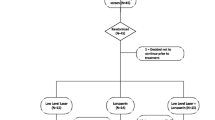
Potential participants were recruited from the local community of Rochester, MN, between September 2016 and August 2017. This report is based on all participants who consented to study, passed study criteria, and went on to be treated in study. All study visits and treatments took place in Mayo Clinic—Rochester Campus. The study adheres to the consort guidelines on reporting clinical trials [10] as demonstrated in the consort diagram presented in Fig. 1.

participant flow in study from first study contact to last study contact
Participants
Eligible subject had to have a BMI between 25.0 and 29.9 kg/m2 and be motivated to lose weight. All interested individuals called a central number and underwent a 10-min phone pre-screen. If they passed the telephone pre-screen, they were invited to attend an in-person consent visit. They were excluded if they did not meet study entry criteria which included criteria such as being under the age of 18, not meeting BMI criteria of 25–29.9 kg/m2, not consenting to study, being on a concurrent weight loss program (including weight-loss medicines or exercise programs), having a weight fluctuation of 20 lb or more in past 6 months, having a medical unstable condition, having a positive pregnancy test, having an infection or having had surgery or other external trauma in the area to be treated by the laser, having a condition that affects weight levels, having a significant psychiatric condition such as depression (as measured by the CESD-R), being photosensitive, or having any other condition which may hinder participation or adherence to study procedures.
After participants consented to participation, they signed a written informed consent and were screened for study eligibility. If they passed the post-consent screening procedures, they were invited to participate. If they accepted the invitation, they were assigned the next available subject ID number and allocated to the appropriate treatment group using the pre-prepared randomization envelopes. Participants attended their in-person study visits every week at approximately the same time (± 2 h) and same day of the week.
Randomization
A computer-generated randomization schedule was prepared by the study statistician using blocks of size N = 6 to ensure that after every sixth participant was randomized and an equal number of subjects was assigned to each treatment group. Using this randomization schedule, sealed randomization envelopes were prepared by administrative staff within the Division of Biomedical Statistics and Informatics. These randomization envelopes were labeled according to subject ID number and contained an index card indicating the treatment assignment for the given subject.
Interventions
LLLT—The LLLT device consists of a multiple-head low-level diode laser with six independent diode laser heads. Each diode emits 532 nm (green) laser light. In the active device (Erchonia1 LipoLaser; Erchonia Medical, Inc., McKinney, TX) [11], each diode generates a 17-mW output. The average number of treatments can vary, depending on the adipose makeup on the patient [12]. In this trial, subjects underwent 12 treatments of LLLT at varying frequencies (1, 2, or 3 times per week). The laser used in this study was an Erchonia® Zerona™ v2.0 Laser. This LLLT has been approved by the Food and Drug Administration (FDA) as a non-invasive dermatological esthetic treatment to reduce the circumference of hips, waist, and thighs. Lasers were targeted at central adiposity in the abdomen region for 30 min and the central region of their back for another 30 min. The lasers were placed approximately 6 in. from the targeted area during the treatment cycle and each treatment occurred within 48 h to 7 days apart.
The Erchonia® Zerona™ 2.0 Laser (used in this study) has been approved by the FDA (K123237) as a non-invasive dermatological esthetic treatment which can be used by individuals intending to reduce circumference of hips, waist, and thighs. Justification for the assertion of anticipated safety and effectiveness of the Erchonia® Zerona 6 Headed Scanner (EZ6) for application to reducing body circumference is found through several FDA clearances for Erchonia® Low Level Laser devices for body circumference reduction indications. For all of the 510(k) clearances, the assigned Product Code is OLI, defined as follows:
Device: fat-reducing low-level laser
Regulation description: low-level laser system for esthetic use
Definition: non-invasive reduction in fat layer for body contouring
Technical method: use of low-level laser energy to create pores in adipocyte cells to release lipids (triglycerides)
Target area: adipocyte cells within the subcutaneous fat layer of the body; this could include abdomen (waist), thighs, and hips
Under 21 CFR 878,5400, the FDA identifies this generic type of device as: “A Low Level Laser System for Aesthetic Use is a device using low level laser energy for the disruption of adipocyte cells within the fat layer for the release of fat and lipids from these cells for non-invasive aesthetic use.”
The procedure administration protocol for each session was as follows:
The study participant is correctly fitted with the safety glasses.
The participant lies comfortably flat on his or her back on the procedure table such that the front area of the subject’s body is facing upwards.
The Erchonia® Zerona 6 Headed Scanner (EZ6) diodes are positioned at a distance of 6 in. above the subject’s lower and upper abdomen, stomach, centered along the body’s midline (the “line” that vertically “dissects” the body into two equal halves).
The Erchonia® Zerona 6 Headed Scanner (EZ6) is then activated for 30 min over the subject’s anterior (frontal) region. Each scanner emits to the subject a laser beam of approximately 17 mW with a wavelength of 532 nm, and creates a spiraling circle pattern that is totally random and independent from the others. These patterns overlap each other to guarantee total coverage within the target area of approximately 80 in.2/516 cm2.
The participant then turns over to lie flat on his or her stomach such that the posterior treatment area of the subject’s body encompassing the region spanning from the participant’s back down though the central body region is facing upwards.
The Erchonia® Zerona 6 Headed Scanner (EZ6) diodes are positioned at a distance of 6 in. above the posterior treatment area, centered along the body’s midline, the same as for the anterior region.
The Erchonia® Zerona 6 Headed Scanner (EZ6) is then activated for 30 min over the subject’s posterior region. Each scanner emits to the subject a laser beam of approximately 17 mW with a wavelength of 532 nm over 15 min, and creates a spiraling circle pattern that is totally random and independent from the others. These patterns overlap each other to guarantee total coverage within the target area of approximately 80 in.2/516 cm2. This converts to each patient receiving approximately 59.2 MJ/cm2 in energy density in the front and the same amount in the back, per session.
The participant’s safety glasses are removed and the procedure administration session is over.
An Erchonia expert trained the study staff in-person on the use and maintenance of the LLLT on December 12, 2013 for a prior study utilizing LLLT. This training session has been documented. They were available for on-the-spot questions and for any online retraining, as needed. The study coordinators were under the direction of the nurse coordinator/supervisor (DFR).
Behavioral intervention—Upon study entry, the study coordinator completed a brief (10-min) individual behavioral intervention introducing and reviewing a patient education resource, the Mayo Clinic “My Weight Solution©” manual, with the study participant, and a copy was given to them to keep. Topics in this manual included motivational strategies, social support, goal-setting strategies, nutritional recommendations, and strategies for physical activity.
Outcomes and safety measures
The primary outcomes included (1) 3% body weight loss from baseline; (2) anthropometric measures of waist (WC) and hip circumference; (3) body composition measurements via InBody 770, a bioelectrical impedance (BIA) scale used to measure participant weight, height, and body composition analysis (intracellular water, extracellular water, dry lean mass, body fat mass), and estimate visceral fat [13]; (4) linear analogue self-assessment (LASA) to self-assess quality of life (QoL) [14,15,16]; (5) participant motivation to reduce weight, self-assessed at baseline prior to study interventions; (6) Body Areas Satisfaction Scale (BASS), a subscale from the Multidimensional Body-Self Relations Questionnaire (MBSRQ) [17] used to self-assess participants’ self-perceived body image and satisfaction of eight specific body areas [18, 19]; (7) Body Appreciation Scale (BAS), a scale to self-assess positive body image [20]; and (8) adherence to the study interventions recorded by staff as attendance to the laser treatments.
The safety measures included (1) adverse events, (2) concomitant medication, (3) self-reported depression using the Center for Epidemiologic Studies Depression Scale Revised (CESD-R), and (4) urine pregnancy tests.
Study schedule
Study visits were divided into three phases: screening (09/30/16 to 08/27/17), treatment (10/04/16 to 11/30/17), and post-treatment (11/11/16 to 03/05/18). All participants were in study for 6 months. The screen phase included a pre-screen phone interview and a combined consent/screen/randomization visit. Once randomized, participants entered the treatment phase based on their randomization schedule. If randomized to group A—the participant reported for treatment for 4 sequential weeks (3 times per week); if randomized to group B—the participant reported for treatment for 6 sequential weeks (2 times per week); if randomized to group C—the participant reported for treatment for 12 sequential weeks (1 time per week). One week after the last treatment, participants received a safety phone contact. Participants were required to complete an in-person study visits at weeks 4, 6, 12, and 26 during which time the study staff collected data pertaining to QoL (LASA), body image (BAS and BASS), safety (adverse events and concomitant medication), as well as vitals, BIA, and body measurements. In addition, during the final visit (week 26), satisfaction was measured via an end-of-study self-assessment survey.
Statistical analysis
The primary endpoint for this investigation was change in WC from baseline, and for this endpoint a reduction of 1.0 cm was considered clinically meaningful. Based on preliminary data from our previous study [5], we determined that for this randomized phase II study, a sample size of N = 20 per group would provide statistical power (one-tailed, alpha = 0.20) of approximately 80% to assess whether additional studies are warranted [21].
Data were summarized using mean ± SD for continuous variables and frequency percentages for nominal variables. Anthropometric measures at each of the study visits (weeks 4, 6, 12, and 26) were compared to baseline using the paired t test and compared across treatment groups using analysis of covariance with the baseline value included as the covariate. Three pairwise treatment group comparisons were of specific interest: the change in WC from baseline to week 4 was compared between those receiving LLLT three per week and those receiving LLLT twice and once per week; and the change in WC from baseline to week 6 was compared between those receiving LLLT twice per week and those receiving LLLT once per week. The results of these comparisons are summarized by presenting the effect estimate along with 90% confidence intervals and one-tailed p values. All other measures were analyzed using two-tailed tests. If the overall comparison across treatment groups was statistically significant at the p < 0.05 level, linear contrasts were used to perform pairwise treatment group comparisons. Data were managed using the REDCap tool hosted at Mayo Clinic [22], and analyses were conducted using SAS statistical software [23].
Results
Study recruitment included internet postings (75%), wait lists (14%), word of mouth (8%), and internal flyers (3%). Of the 245 participants pre-screened, 60 were consented and randomized (20 per group) (Fig. 1). All participants but one (98%) completed at least 10 of the 12 treatments. For group A, 18 (90%) participants completed all 12 sessions and 2 (10%) completed 11 sessions. For group B, 17 (85%) participants completed all 12 sessions, 2 (10%) completed 11 sessions, and 1 (5%) completed 10 sessions. For group C, 18 (90%) participants completed all 12 sessions, 1 (5%) completed 11 sessions, and 1 (5%) completed 7 sessions (this last participant had to terminate the treatments early due to pregnancy; see details below).
The majority of participants were white (95%), female (90%), married/living as married (72%), with at least a 4-year college degree (53%), average age of 43.7 years (± 9.2), and moderately active (72%). No between-group differences in concern, motivation, importance, and confidence in weight loss, as well as stress level were detected using visual analogue scales (0 to 10, with 0 being the lowest and 10 being the highest) (see Table 1).
Body anthropometric and composition measurements are summarized in Table 2, and the percentage of participants who had weight loss of ≥ 3% is summarized in Table 3. In those receiving LLLT 3 times per week for 4 weeks (group A), there were no significant reductions from baseline in weight, BMI, or waist circumference at any time point. In those receiving LLLT 2 times per week for 6 weeks (group B), weight, BMI, and waist circumference were all significantly reduced from baseline at weeks 4 and 6, with significant reductions in waist circumference also observed at 12 and 26 weeks. For those receiving LLLT once per week for 12 weeks (group C), waist circumference was found to be significantly reduced from baseline at week 6. For group A, no significant changes in body composition measurements from baseline were detected at any time periods. For groups B and C, significant reductions from baseline were observed starting at week 4 for body fat mass, percent body fat, and visceral fat. When changes from baseline were compared across groups, the only significant finding was for the change in body fat mass at week 6, with group B experiencing a larger reduction compared to group A.
In order to assess whether an increased frequency of LLLT was associated with a larger reduction in body circumference, one-tailed tests were used for three comparisons of specific interest including the change in WC from baseline to week 4 for group A compared to group B and group A compared to group C as well as the change in WC from baseline to week 6 for group B versus group C. In all cases, no meaningful differences were found (week 4—A vs. B effect estimate = + 0.62, 90% CI − 0.68 to + 1.92, one-tailed p = 0.783; week 4—A vs. C effect estimate = − 0.50, 90% CI − 1.80 to + 0.80, one-tailed p = 0.266; week 6—B vs. C effect estimate = − 0.40, 90% CI − 1.98 to + 1.18, one-tailed p = 0.340). When all other measurements were compared across groups, reductions in weight and BMI at 6 weeks were found to be statistically significant with those in group B having significantly larger reductions compared to group A (Table 3).
No significant differences were observed across groups with respect to changes from baseline in motivation, body satisfaction, body appreciation and overall quality of life (QoL) (Table 4), except for the QoL domain of physical, which was significant at week 12 for group B. However, changes across time show a positive trend in BASS (weeks 6, 12, and 26), BAS (weeks 6, 12, and 26), motivation to lose weight, overall QoL (weeks 6 and 12), and physical QoL (weeks 6, 12, and 26) in group B compared to baseline.
One serious adverse event of pregnancy was reported after the seventh treatment in a participant in group C. All treatments were stopped and the patient was followed through a healthy delivery. During the course of the 26-week study, 29 (48%) of the participants reported 39 adverse events, none of which were determined to be related to the LLLT. Depression was monitored throughout the study using the CESD-R with three participants reporting situational depression at some point during the study. Upon further assessment, the situational depression was found not to be related to the study and the patient was cared for in the appropriate manner per appropriate medical care. These participants continued in the study and treatment was not halted.
At the end of our study, participants were asked questions about changes made in lifestyle (e.g., reducing calorie intake, making healthier nutrition choices, exercising, increasing water intake, wearing body-constricting undergarments) during the 26-week study period. The percent of patients reporting lifestyle changes was similar across groups with 50%, 45%, and 60% reporting one or more lifestyle changes in groups A, B, and C, respectively. In addition, no significant difference was observed between groups with respect to satisfaction with treatment assignment (overall satisfaction with the frequency of LLLT was reported as “satisfied” or “extremely satisfied” at week 26 (end of study) by 50% (10/20) in group A, 65% (13/20) in group B, and 40% (8/20) in group C) (Table 5).
Discussion
In this pilot project, three frequencies of LLLT treatment therapy were examined and results indicate that treating adults twice weekly for 6 weeks (group B) was the best option for adults who are overweight and motivated to lose weight compared to once a week (group C) or 3 times a week (group A).
For group B (2 LLLTs per week for 6 weeks), a significant reduction was noted when compared to group A (3 LLLTs per week for 4 weeks) in weight and BMI; within groups, a significant reduction from baseline was noted in group B for weight, BMI, body fat mass, and percent body fat at weeks 4 and 6, as well as a reduction in visceral fat level at weeks 4, 6, and 12; a reduction was noted in waist circumference at weeks 4, 6, 12, and 26; a significant waist-to-hip ratio reduction was noted at week 12; improvement in body area satisfaction and body appreciation was noted at weeks 6, 12, and 26 and improvement in overall quality of life at weeks 6 and 12. No side effects were reported related to LLLT in any of the three treatment conditions. Overall satisfaction was higher among participants assigned to twice-weekly treatments (group B) than the other groups. For group B, 25% of the patients achieved 3% weight loss at end of the treatments (week 6) and 15% continued to maintain the 3% weight loss from baseline at end of study (week 26).
The finding that participants receiving two LLLTs per week for 6 weeks (group B) experienced greater reduction in weight and body fat mass at 6 weeks compared to those receiving three LLLTs per week for 4 weeks (group A) suggests that a longer duration of moderate intensity is preferable when compared to a short duration of higher-intensity therapy. In addition, no evidence was observed to suggest that 12-week outcomes were improved with 12 weeks of once-weekly LLLT (group C) compared to 6 weeks of twice-weekly LLLT (group B). Observed differences at 12 weeks suggest that the moderate intensity (2 LLLTs per week for 6 weeks—group B) is preferred.
Consistent with previous literature, LLLT was associated with reduction in weight, BMI, WC, waist-to-hip ratio, and body fat mass. Participants in this pilot project demonstrated that these effects may be ideally achieved when they are delivered twice weekly. This association is confirmed by another study of 67 participants who were overweight and were randomized to six LLLT treatments of either true LLLT or sham LLLT [6]. Within this placebo-controlled study, 63% of the active participants (vs. 6% in the sham arm) lost a combined total of ≥ 3.0 in. from waist, hip, and bilateral thighs from baseline to end of treatment. In our study, we observed a loss of 2.4 in. in waist circumference at end of treatment (compared to baseline) and a reduction of 1.5 in. at end of study compared to baseline. It is important to note that retrospective studies have supported the body measurement reductions achieved (e.g., waist, hips, thighs) with LLLT and reported concomitant decreases in weight [7].
The current US guidelines for clinical weight management recommend a 6-month weight loss goal of 3–5% from baseline weight [24]. Even such a modest weight loss can produce health benefits through reductions in triglycerides, blood sugars, and risk for diabetes, with larger weight loss producing greater benefits. In this study, all groups were able achieve this, but group B had the most consistent weight loss throughout the study. We propose that LLLT 2 times per week for 6 weeks achieves an appropriate balance between intensity and duration with the LLLT protocol. We were not able to find any prior published studies focused on these weight loss goals for patients who were overweight with a BMI of 25–29.9 kg/m2. Past studies have focused on patients who are obese and have used medications (i.e., orlistat, metformin, naltrexone/bupropion, lorcaserin) or surgical interventions with other lifestyle modifications (i.e., behavioral interventions). In these studies, a 5% weight loss from baseline has been achieved in 33% to 80% [5, 24,25,26,27] of participants, but whether such a loss has been able to be maintained long term is unknown.
Low QoL has been associated with obesity and overweight [28, 29]. Indeed, one past study compared QOL of individuals who were overweight and obese to the general population and observed them to have lower QOL, especially in the physical and mental domains. With weight loss, these QoL domains improved [28]. Other studies have observed that QoL domains most improved with weight loss are the physical and mental domains [28, 30,31,32,33,34]. In the present study, we observed that LLLT in group B was associated with improvements in overall QoL. Specifically, we observed significant improvement in the physical role domain of QOL at weeks 6, 12, and 26 (+ 0.8 ± 1.1, + 1.0 ± 1.1, and + 0.7 ± 1.1, respectively). We observed that participants had an improved attitude toward their body (better appreciation and satisfaction) as well as improved motivation after starting LLLT. These changes were insignificant for group B in which the trend continued until the end of the study.
Across the three groups, 55–65% of the participants reported that LLLT was helpful and 35–60% said they would recommend it to friends. Participants may have set unrealistic expectations of a larger weight loss with LLLT treatment and became disappointed when those expectations were not met. This mind set phenomenon is not unusual in weight loss programs where setting unrealistic expectations is a barrier to weight loss [35]. In a study of 60 females who are obese (mean BMI of 36.3 kg/m2) in which participants set a goal of 32% body weight reduction, and the average weight loss was less than 17% from their baseline weight, participants deemed the program a failure [36]. Two other studies with individuals with BMIs ≥ 40 kg/m2 or higher observed this same phenomenon [37, 38]. Individuals with higher baseline weight may have more unrealistic goals for weight loss and high unrealistic expectations set prior to the program which led to poor program compliance and worse outcomes [35]. In the current investigation, study staff provided accurate information concerning LLLT and healthy weight loss expectations (5–10% per US guidelines [24]). This discussion may have improved adherence and reduced study attrition. In a prior study of 1785 patients who were obese throughout 23 Italian medical centers, higher weight loss expectations were associated with higher 12-month attrition [39]. In addition, a recent review of 19 weight loss studies observed that higher stages of change at baseline, higher initial weight loss, higher education, and older age are predictors of adherence to weight loss interventions [40]. Future studies should include counseling components to manage expectations of weight loss.
Finally, all but one prior LLLT studies have focused their outcome measures of weight, BMI, and anthropometric measurements at end of treatment and not on long-term (6 months) outcomes. We have evaluated outcomes 3 months after LLLT therapy completion [5]. In that study, we observed that patients receiving LLLT had no weight changes (− 1.4 ± 3.6 kg) and waist circumference lost centimeters (− 2.8 ± 4.3 cm) within 3 months of the last laser treatment [5]. In the current study, we observed that as motivation decreased post-treatment, the success made toward weight loss and reduced waist circumference diminished and weight and inches were regained. In order for weight loss maintenance to be successful, it should be maintained for 2 to 4 years [41] and, to date, the only successful programs have been ones which consist of high physical activity, low-calorie/low-fat diets, and daily self-monitoring [24, 41, 42]. This can only be achieved through motivational enhancement. More studies are needed to determine the long-term maintenance efficacy of LLLT.
Although our small sample size is acceptable for a pilot study, it limits the ability to detect significant differences between groups. In addition, the open-label design limits our study due to patient selection bias [43], participant retention bias [44], and participant performance bias [45]. Some eligible participants decided not to participate knowing there was a chance they would not receive their preferred schedule. Since LLLT is a new weight loss intervention, it is not known what healthy lifestyle recommendations can be tailored to LLLT, such as more focus on strength training or nutritional guidelines. We provided all participants with a self-help guide on weight loss at study enrollment and a brief orientation to the manual. We did not provide any further reference to the manual during study participation or any counseling thereafter. Fifty-five percent of the participants indicated the counseling manual was helpful but would have welcomed behavioral intervention throughout the study. A strengthened behavioral modification component could be explored as a potential co-intervention to LLLT for future studies.
Conclusions
Providing 12 LLLT treatments over the course of 6 weeks twice per week is effective for reducing body weight, BMI, body fat mass, and percent body fat as well as improving body satisfaction, body appreciation, and overall quality of life. At end of treatment, 30% of participants who were overweight (BMI of 25.0 to 29.9 kg/m2) in the twice-weekly group lost greater than 3% of baseline weight. More research should be conducted to determine if additional improvements in body anthropometric measurements can be achieved by adding other non-burdensome components to the LLLT intervention such as behavioral counseling.
Availability of data and materials
All data supporting the study findings are contained within this manuscript and within file NCT02877004 on www.ClinicalTrials.gov.
Abbreviations
- BIA:
-
bioelectrical impedance
- BAS:
-
Body Appreciation Scale
- BASS:
-
Body Areas Satisfaction Scale
- BMI:
-
body mass index
- CESD-R:
-
Center for Epidemiology Studies Depression Scale—Revised
- LASA:
-
Linear Analogue Self-Assessment
- LLLT:
-
low-level laser therapy
- MBSRQ:
-
Multidimensional Body-Self Relations Questionnaire
- QoL:
-
quality of life
- WC:
-
waist circumference
References
Prevalence of overweight, obesity, and extreme obesity among adults: United States, trends 1960–1962 through 2007–2008 [http://www.cdc.gov/NCHS/data/hestat/obesity_adult_07_08/obesity_adult_07_08.pdf] Accessed 01 Nov 2011
Avci P, Nyame TT, Gupta GK, Sadasivam M, Hamblin MR (2013) Low-level laser therapy for fat layer reduction: a comprehensive review. Lasers Surg Med 45(6):349–357
Neira R, Arroyave J, Ramirez H, Ortiz CL, Solarte E, Sequeda F, Gutierrez MI (2002) Fat liquefaction: effect of low-level laser energy on adipose tissue. Plast Reconstr Surg 110(3):912–922 discussion 923-915
Chen AC, Arany PR, Huang YY, Tomkinson EM, Sharma SK, Kharkwal GB, Saleem T, Mooney D, Yull FE, Blackwell TS et al (2011) Low-level laser therapy activates NF-kB via generation of reactive oxygen species in mouse embryonic fibroblasts. PLoS One 6(7):e22453
Croghan IT, Ebbert JO, Schroeder DR, Hurt RT, Hagstrom V, Clark MM (2016) A randomized, open-label pilot of the combination of low-level laser therapy and lorcaserin for weight loss. BMC Obes 3:42
Jackson RF, Roche GC, Shanks SC (2013) A double-blind, placebo-controlled randomized trial evaluating the ability of low-level laser therapy to improve the appearance of cellulite. Lasers Surg Med 45(3):141–147
McRae E, Boris J (2013) Independent evaluation of low-level laser therapy at 635 nm for non-invasive body contouring of the waist, hips, and thighs. Lasers Surg Med 45(1):1–7
Jackson RF, Stern FA, Neira R, Ortiz-Neira CL, Maloney J (2012) Application of low-level laser therapy for noninvasive body contouring. Lasers Surg Med 44(3):211–217
Nestor MS, Zarraga MB, Park H (2012) Effect of 635nm low-level laser therapy on upper arm circumference reduction: a double-blind, randomized, sham-controlled trial. J Clin Aesthet Dermatol 5(2):42–48
Consort: Transparent Reporting of Trials [www.consort-statement.org/consort-statement/flow-diagram] Accessed 15 Aug 2016
Zerona [http://www.myzerona.com/professional] Accessed 01 Sept 2011
Mulholland RS, Paul MD, Chalfoun C (2011) Noninvasive body contouring with radiofrequency, ultrasound, cryolipolysis, and low-level laser therapy. Clin Plast Surg 38(3):503–520 vii-iii
InBody 770: Premium Solution for Your Health [http://www.e-inbody.com/global/product/InBody770.aspx] Accessed 15 March 2016
Singh JA, Satele D, Pattabasavaiah S, Buckner JC, Sloan JA (2014) Normative data and clinically significant effect sizes for single-item numerical linear analogue self-assessment (LASA) scales. Health Qual Life Outcomes 12(1):187
Hyland M, Sodergren S (1996) Development of a new type of global quality of life scale, and comparison of performance and preference for 12 global scales. Qual Life Res 5(5):469–480
Locke DE, Decker PA, Sloan JA, Brown PD, Malec JF, Clark MM, Rummans TA, Ballman KV, Schaefer PL, Buckner JC (2007) Validation of single-item linear analog scale assessment of quality of life in neuro-oncology patients. J Pain Symptom Manag 34(6):628–638
Cash TF (2015) Multidimensional body–self relations questionnaire (MBSRQ). In: Wade T (ed) Encyclopedia of feeding and eating disorders. Springer, Singapore
Cash T (1994) The multidimensional body-self relations questionnaire user’s manual. Norfolk
Clark MM, Croghan IT, Reading S, Schroeder DR, Stoner SM, Patten CA, Vickers KS (2005) The relationship of body image dissatisfaction to cigarette smoking in college students. Body Image 2(3):263–270
Avalos L, Tylka TL, Wood-Barcalow N (2005) The body appreciation scale: development and psychometric evaluation. Body Image 2(3):285–297
Rubinstein LV, Korn EL, Freidlin B, Hunsberger S, Ivy SP, Smith MA (2005) Design issues of randomized phase II trials and a proposal for phase II screening trials. J Clin Oncol 23(28):7199–7206
Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG (2009) Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 42(2):377–381
SAS Institute Inc (2017) SAS/STAT User’s Guide—Version 9.4. SAS Institute, Cary, NC
Jensen M, Ryan D, Apovian C, Ard J, Comuzzie A, Donato K, Hu F, Hubbard V, Jakicic J, Kushner R et al (2014) 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults. J Am Coll Cardiol 63(25 Part B):2985–3023
Ornellas T, Chavez B (2011) Naltrexone SR/bupropion SR (Contrave): a new approach to weight loss in obese adults. P&T 36(5):255–262
Fidler MC, Sanchez M, Raether B, Weissman NJ, Smith SR, Shanahan WR, Anderson CM (2011) A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab 96(10):3067–3077
O'Neil PM, Smith SR, Weissman NJ, Fidler MC, Sanchez M, Zhang J, Raether B, Anderson CM, Shanahan WR (2012) Randomized placebo-controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM study. Obesity 20(7):1426–1436
Blissmer B, Riebe D, Dye G, Ruggiero L, Greene G, Caldwell M (2006) Health-related quality of life following a clinical weight loss intervention among overweight and obese adults: intervention and 24 month follow-up effects. Health Qual Life Outcomes 4:43
Hajian-Tilaki K, Heidari B, Hajian-Tilaki A (2016) Solitary and combined negative influences of diabetes, obesity and hypertension on health-related quality of life of elderly individuals: A population-based cross-sectional study. Diabetes Metab Syndr 10(2 Suppl 1):S37-42.
Batsis JA, Clark MM, Grothe K, Lopez-Jimenez F, Collazo-Clavell ML, Somers VK, Sarr MG (2009) Self-efficacy after bariatric surgery for obesity. A population-based cohort study. Appetite 52(3):637–645
Batsis JA, Lopez-Jimenez F, Collazo-Clavell ML, Clark MM, Somers VK, Sarr MG (2009) Quality of life after bariatric surgery: a population-based cohort study. Am J Med 122(11):1055 e1051–1055 e1010
Rothberg AE, McEwen LN, Kraftson AT, Neshewat GM, Fowler CE, Burant CF, Herman WH (2014) The impact of weight loss on health-related quality-of-life: implications for cost-effectiveness analyses. Qual Life Res 23(4):1371–1376
Pan A, Kawachi I, Luo N, Manson JE, Willett WC, Hu FB, Okereke OI (2014) Changes in body weight and health-related quality of life: 2 cohorts of US women. Am J Epidemiol 180(3):254–262
Leon-Munoz LM, Guallar-Castillon P, Banegas JR, Gutierrez-Fisac JL, Lopez-Garcia E, Jimenez FJ, Rodriguez-Artalejo F (2005) Changes in body weight and health-related quality-of-life in the older adult population. Int J Obes 29(11):1385–1391
Lagerros YT, Rossner S (2013) Obesity management: what brings success? Ther Adv Gastroenterol 6(1):77–88
Foster GD, Wadden TA, Vogt RA, Brewer G (1997) What is a reasonable weight loss? Patients’ expectations and evaluations of obesity treatment outcomes. J Consult Clin Psychol 65(1):79–85
Linne Y, Hemmingsson E, Adolfsson B, Ramsten J, Rossner S (2002) Patient expectations of obesity treatment—the experience from a day-care unit. Int J Obes Relat Metab Disord 26(5):739–741
Foster GD, Wadden TA, Phelan S, Sarwer DB, Sanderson RS (2001) Obese patients’ perceptions of treatment outcomes and the factors that influence them. Arch Intern Med 161(17):2133–2139
Dalle Grave R, Calugi S, Molinari E, Petroni ML, Bondi M, Compare A, Marchesini G, Group QS (2005) Weight loss expectations in obese patients and treatment attrition: an observational multicenter study. Obes Res 13(11):1961–1969
Leung AWY, Chan RSM, Sea MMM, Woo J (2017) An overview of factors associated with adherence to lifestyle modification programs for weight management in adults. Int J Environ Res Public Health 14(8):922.
Wing RR, Hill JO (2001) Successful weight loss maintenance. Annu Rev Nutr 21:323–341
The Practical Guide: Identification, Evaluation, and Treatment of Overweight and Ovesity in Adults [www.nhlbi.nih.gov/guidelines/obesity/prctgd_c.pdf] Accessed 05 Feb 2019
Juni P, Altman DG, Egger M (2001) Systematic reviews in health care: assessing the quality of controlled clinical trials. BMJ 323(7303):42–46
Halpern SD (2003) Evaluating preference effects in partially unblinded, randomized clinical trials. J Clin Epidemiol 56(2):109–115
Rucker G (1989) A two-stage trial design for testing treatment, self-selection and treatment preference effects. Stat Med 8(4):477–485
Acknowledgments
Special thanks to the exceptional research staff of the Mayo Clinic Department of Medicine Clinical Research Office for their patience and persistence in helping to collect, compile, and organize these data. Special thanks to Bonnie Donelan Dunlap, Donna Rasmussen, and Sara Gifford for all their hard work and dedication to this study. The authors also wish to thank the study participants who participated in this clinical trial, without whom this project would not have been possible.
Funding
The data entry system used was REDCap, supported in part by the Center for Clinical and Translational Science award (UL1 TR000135) from the National Center for Advancing Translational Sciences (NCATS). This funding body had no oversight and did not provide any input or direction to the current study design, collection, analysis or interpretation of data, as well as this report.
Author information
Authors and Affiliations
Contributions
All the authors participated in the study concept and design, analysis and interpretation of data, drafting and revising the paper, and have seen and approved the final version of the manuscript.
I.T.C.—conceived of the study concept and design; obtained funding and provided administrative, technical, and material support; had full oversight of the study conduct during data collection; had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis; and also drafted the manuscript and participated in critical revision of the manuscript for important intellectual content.
J.O.E., R.T.H., M.D.J.—participated in the study design and were responsible for safety screening for study entry and the safety oversight of the study subjects while on study. They also participated in the review and interpretation of study results, and critical revision of the manuscript for important intellectual content.
S.C.F.—reviewed the study flow, screened the study participants, oversaw all the study visits and LLLT treatments, as well as worked with the study clinicians and PI to address patient safety issues and with the PI and statistician to clean out the electronic data as well as provide reviews and edits to the manuscript.
D.R.S.—participated in the study design and was responsible for data quality checks and data analysis; he also had full access to all the data in the study and takes full responsibility for the integrity of the data and the accuracy of the data analysis as well as participating in the manuscript reviews and edits.
M.M.C.—participated in the study design and was instrumental in the development of the patient manual “My Weight Solution.” He provided training to the staff on the brief intervention utilizing this manual and participated in the manuscript review and provided edits.
Corresponding author
Ethics declarations
Ethics and consent to participate
In accordance with the Declaration of Helsinki, this study was reviewed and approved (ID 16-004817) by the Mayo Clinic Institutional Review Board (IRB). Mayo Clinic IRB–approved written informed consent was obtained for all study participants prior to study participation.
Consent to publish
Not applicable.
Competing interests
I.T.C.—no competing interests to declare.
R.T.H.—reports consulting fees from Nestlé, and research funding from NIH and InBody outside the submitted work.
D.R.S.—no competing interests to declare.
S.C.F.—no competing interests to declare.
M.D.J.—no competing interests to declare.
M.M.C.—no competing interests to declare.
J.O.E.—no competing interests to declare.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Croghan, I.T., Hurt, R.T., Schroeder, D.R. et al. Low-level laser therapy for weight reduction: a randomized pilot study. Lasers Med Sci 35, 663–675 (2020). https://doi.org/10.1007/s10103-019-02867-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10103-019-02867-5