
Abstract
This pilot, double-blind, randomized, placebo-controlled study is aimed at evaluating the effectiveness of low-level laser therapy (LLLT) as a complementary treatment to complete decongestive therapy (CDT) treating lymphedema among breast cancer patients for 12 months post-intervention. Study population was breast cancer patients who were diagnosed and referred to lymphedema clinic for CDT. Participants (n = 22) were randomized and assigned into either an active laser intervention group or an inactive laser placebo-control group. Active LLLT was administered to participants twice a week at the beginning of each CDT session. Outcome measures included lymphedema symptoms, symptom distress, and limb volume by an infrared perometer. Participants in the active and placebo laser groups were comparable in demographic and clinical predictors of lymphedema. In comparison with the placebo group (83.3%), significantly fewer participants in the active laser group (55.6%) reported more than one lymphedema symptom (p = 0.012) at 12 months post-intervention. Significantly, more patients in the active laser group (44.4%) reported less than two impaired limb mobility symptoms in comparison with the placebo group (33.3%) at 12 months post-intervention (p = 0.017). The active laser group had statistically significant improvements in symptom distress of sadness (p = 0.005) from 73 to 11% and self-perception (p = 0.030) from 36 to 0% over time from baseline to 12-months post-intervention. There was no significant reduction in limb volume. Findings of the trial demonstrated significant benefits of complementary LLLT for relieving symptoms and improvement of emotional distress in breast cancer patients with lymphedema.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lymphedema is a chronic and progressive condition that is characterized by the accumulation of protein-rich lymph fluid in the interstitial spaces [1,2,3]. Lymphedema after breast cancer treatment is a result of damage to the axillary lymph drainage routes caused by surgery or radiation treatment [4] and can develop at any age and at any time [5]. Lymphedema can lead to chronic inflammation, fibrosis, and repeated infection [1,2,3]. Currently there are no medical or surgical cures for lymphedema [6]. Breast cancer survivors with lymphedema may suffer from multiple lymphedema symptoms, including swelling, heaviness, firmness, tenderness, pain, numbness, aching, and stiffness [7,8,9,10]. Consequences of these symptoms include physical deficits involving limb weakness and diminished range of motion limiting an individual’s ability to perform activities of daily living and work duties [8, 11, 12], psychological morbidity [1, 2, 8, 11], decreased quality of life (QOL) [1, 13, 14], and for the remainder of their lifetime increases the risk of infection in the involved extremity [1, 10,11,12, 15].
Complete decongestive therapy (CDT) has been the standard treatment for lymphedema [16,17,18]. CDT is a combination of therapies, including manual lymphatic drainage (MLD), multilayer bandaging (MLB), compression, hygiene and skin care, and modified exercise [1, 2, 15, 16, 18]. Daily and life-long self-management of lymphedema is imperative to prevent worsening of the condition and repeated infections and to ensure patients’ QOL [17,18,19]. Studies have shown that components of CDT are effective in achieving initial reductions in swelling [20, 21], decreasing symptoms [22], and improving QOL [16, 22]. Due to the chronic nature of the condition, maintaining the benefits gained during a course of CDT can be challenging for individuals over time [2].
Low-level laser therapy (LLLT) is a conservative therapy utilizing nonionizing light sources that has been used in the treatment of lymphedema for breast cancer patients [4]. Studies have shown benefits using LLLT in this population [23,24,25,26,27], with proposed mechanisms of action supporting use of this device on human tissues. The basic mechanism for the use of low-level light for therapeutic benefit includes light absorption, targeting and activating cytochrome c oxidase in the mitochondrial membrane, leading to greater consumption of cellular oxygen and ultimately increased metabolic energy production [28], which is used for cellular repair. Research on the use of this laser at low-intensity wavelengths suggest effects at the cellular level [29] with increased lymphangiogenesis [30] and stimulation of lymphatic motoricity [31, 32] facilitating removal of excess protein-rich fluid, and stimulation of macrophage cells and the immune system [30, 33] which decrease the risk of infection. Studies have reported LLLT to be effective in softening fibrous tissue [4, 32] and surgical scars [34], reducing pain [4, 34], reducing arm volume [34,35,36], and improving QOL in the breast cancer population [33]. Long-term benefits have been documented in the literature using LLLT in this population in limited studies [32, 33, 35, 36], with a 12-month follow-up in one study [35], and a 2.5 year follow-up in one study [32]. In November 2006, the Food and Drug Administration approved the use of the RianCorp LTU-904 low-level laser (LLL) for treatment of post-mastectomy lymphedema [37]. Published studies on the effectiveness of LLLT are currently limited due to lack of research design involving double-blind and placebo-control randomization. In addition, there are limited published studies on the effectiveness of LLLT as a complementary treatment to the standard care of CDT for breast cancer survivors. Thus, the purpose of the study was to evaluate whether the use of LLLT as a complementary treatment to CDT would result in additional improvement and long-term benefits to individuals with lymphedema. The primary outcomes were lymphedema symptoms, limb volume, and QOL. We hypothesized that more patients in the active laser group would have fewer symptoms and less symptom distress, better limb volume, and greater treatment adherence than patients in the inactive laser group.
Materials and methods
This study was approved by the Institutional Review Board (IRB) of a larger metropolitan health system and was registered as clinical trial #10-00923. Physicians and nurse practitioners referred potential participants from March 2011, to June 2016. To be enrolled in the trial, potential participants who were willing to participate in the trial had to (1) meet the inclusion and exclusion criteria (Table 1) and (2) obtain written approval from their referring physician, who agreed to allow the patient to participate in the trial.
Study design
A double-blind, randomized, placebo-controlled design was used for this pilot study to explore the effectiveness of LLLT on lymphedema as a complementary intervention to CDT when compared with CDT treatment alone. The primary outcomes were lymphedema symptoms, limb volume, and QOL. We hypothesized that patients in the active laser group would have significantly fewer symptoms, better limb volume, and improved QOL in comparison with those in the inactive laser group.
Participants
Oncologists affiliated with the medical center were informed about the trial. Women who came to the clinic for a lymphedema evaluation were offered a study flyer for participation into the trial. Fifty-four women who met the inclusion criteria were identified. Twenty-two women agreed to participate in the study. Informed consent was obtained from all participants, and subjects were randomized into either an active laser group or an inactive laser group. One subject consented to the study and was randomized, then changed her mind before data collection was performed, so a total of twenty-one women participated in the study (Fig. 1).

Study flow chart
Randomization and blindness
After participants signed the informed consent, they were randomized and assigned into either an active laser intervention group (laser group) or an inactive laser placebo-control group (placebo group) using a non-stratified permuted-block design. Varying block sizes were used to minimize the possibility of predicting the allocation sequence. The study’s statistician, who was not involved in treatment intervention or data collection, performed the randomization. Participants were blinded to the laser allocation. Lymphedema therapists and research assessors who collected outcomes data were also blinded to the randomization of group allocation, as well as to the allocation of the active and inactive laser devices. All clinicians who administrated the laser therapy were blinded to the allocation of the lasers. This was made possible because both the active and inactive laser device presented the same red light during the treatment.
Treatment description
Since CDT has been the standard treatment for breast cancer-related lymphedema (BCRL) [6], all study participants received CDT in this trial. The detailed description of CDT can be found in Table 2. The use of LLL (the LTU-904 laser by RianCorp) was added to CDT. The study utilized a total of 4 lasers, 2 active and 2 inactive laser devices, which were pre-calibrated to provide either active or placebo laser treatment. This information was blinded to participants, lymphedema therapists, and researchers who evaluated outcome measures. Lymphedema therapists were trained in the application of the LLL, and complementary LLLT was administered twice a week at the beginning of each lymphedema treatment session before CDT.
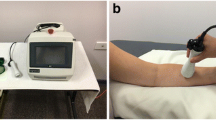
LLLT was performed using a standardized fabric treatment grid made from cut fabric sheets. The grid was pre-fashioned with 40 punctured holes as marked treatment sites, and the holes were divided into four color-coded groups: red, blue, green, and black. The holes alternated colors to allow for spacing of ≥ 2 cm between consecutive same-colored holes. Participants were given their own fabric treatment grid to be used for the duration of the study. The fabric grid was draped over the underarm and medial to lateral chest wall of the affected upper quadrant using pre-determined anatomical landmarks to ensure consistency with each treatment. The laser treatment was applied with the participant positioned in supine with the affected arm abducted to 90° (Fig. 2). The laser was applied over the standardized grid using a corresponding set of color-coded holes alternating each session on a 4-week cycle (Fig. 3). LLLT was applied on the high setting for 1 min at each of 10 sites in the axilla and a portion of the chest wall on the affected side, for a total of 8–16 sessions depending on the time frame needed to achieve clinical goals. The dose per application site was 1.5 J per centimeter squared (J/cm2), for a total dose of 15 J/cm2 delivered at each treatment session.

Laser treatment applied with the patient in supine, affected arm abducted to 90°

Standardized fabric treatment grid and treatment session assignment
Procedures
Members of the research team who were blind to the randomization of the LLL device collected data. Lymphedema therapists were trained in the application of LLLT. Both groups received individualized CDT as determined by their treating therapist. Treatment sessions using LLLT as an adjunct to CDT ranged between 8 and 16 sessions, with the final number of visits for each subject determined by the time needed to achieve maximal circumferential reduction as assessed by girth measurements, and palpation of soft tissue texture/density improvement. Demographic and medical information was obtained at the first visit after evaluation. Data collection of selected outcome measures was performed at the first visit after evaluation, after 8 treatment sessions, after the final treatment session, and at long-term follow-up at 3, 6, and 12 months (Table 3). We followed the research procedures used in our prior studies [38, 39], including use of the perometer as recommended by the manufacturers [38,39,40]. Protection of human subjects was ensured by following the guidelines set forth by the IRB. Each participant signed the written consent to the study.
Clinical and outcome measures
Demographic and medical information
A structured interview tool was used to gather demographic and medical information [38,39,40]. Demographic and medical data collected included age, ethnicity, occupation, education, diagnosis, treatment, nodal status, numbers of lymph nodes removed, co-morbidities, and family and medical history (such as breast cancer history, breast cancer gene status, and family history of lymphedema). Medical information was verified by reviewing participants’ medical records as needed.
Height and BMI
Height was measured without shoes on to the nearest 0.1 cm with a portable stadiometer. An electrical bioimpedance device (InBody 520, Biospace Co., Ltd.) was employed to measure the weight, and the device automatically calculated BMI using the formula: weight (kg)/height (m2) [39].
Lymphedema symptoms and symptom distress
The lymphedema and breast cancer symptom experience index (BCLE-SEI) is a valid, reliable, 5-point Likert-type self-report instrument to assess symptoms related to lymphedema or fluid accumulation [40,41,42]. This instrument consists of two parts, one evaluating the occurrence of lymphedema symptoms and another evaluating QOL in terms of symptom distress. The lymphedema symptom assessment (part 1) assesses impaired limb mobility in the shoulder, arm, elbow, wrist, and fingers. It also assesses arm swelling, breast swelling, chest wall swelling, heaviness, firmness, tightness, stiffness, numbness, tenderness, pain/aching/soreness, redness, blistering, burning, stabbing, tingling (pain and needles), hotness, seroma, and limb fatigue/weakness. Symptom distress (part 2) evaluates the adverse impact and suffering evoked by one’s experience of lymphedema symptoms [40,41,42]. Symptom distress includes dimensions of daily living, social function, sleep disturbance, sexuality, emotional/ psychological distress, and days absent from work.
Infrared perometer measurement of limb volume
Perometry was performed on each arm as it was held horizontally. The Perometer 350S maps a 3-dimensional graph of the affected and non-affected extremities using numerous rectilinear light beams and interfaces with a computer for data analysis and storage. A 3-dimensional limb image was generated and LV was calculated. This optoelectronic method has a standard deviation of 8.9 ml (arm), less than 0.5% of LV with repeated measuring. Procedures for perometry were based on protocol used in other research [21].
Statistical analysis
Demographic and treatment variables
Adherence to the Research Program we defined participants’ adherence to the research program as patients having completed each follow-up visit without loss to follow-up. SPSS version 22 (IBM, Armonk, NY, USA) was used for statistical analysis. Descriptive statistics were calculated for baseline demographic (age, weight, BMI, percentage of body fat, marital status, employment, retirement status, disability) and clinical characteristics (primary cancer stage, secondary cancer stage, mastectomy, lumpectomy, chemotherapy, radiation, auxiliary lymph node treatment, lymph nodes removed, family history of lymphedema) overall and within each treatment arm. All point estimates were generated with 95% confidence intervals. Mean and standard deviations were used to summarize continuous variables; frequencies and percentages were used to summarize categorical variables.
Lymphedema symptoms, limb volume, and QOL
Because even a 5% limb volume increase enables detectable differences in QOL, lymphedema was defined as a perometer measurement of ≥ 5% limb volume increase in the ipsilateral arm in comparison with the changes in the contralateral arm [43]. As the 24 lymphedema symptoms denote clusters of symptoms, i.e., groups of more than two symptoms that occur together, to indicate different biological mechanism [43], we categorized the 24 symptoms into three identifying symptom clusters of impaired limb mobility (limb mobility in the shoulder, arm, elbow, wrist, and fingers, tightness and stiffness), fluid accumulation symptoms (arm swelling, breast swelling, chest wall swelling, seroma, heaviness, and firmness), and pain/discomfort symptoms (pain/aching/soreness, numbness, tenderness, redness, blistering, burning, stabbing, tingling, hotness, limb fatigue/weakness).
Distributions of the outcome variables, including (a) limb volume by perometer ≥ 5%, (b) count of lymphedema symptoms (0 vs 1–24 symptoms), (c) symptom clusters (reporting 2 or more symptoms related to impaired limb mobility, fluid accumulation, and pain/discomfort), and (d) symptom distress, were summarized at each time point (visit 1 to visit 6) and associations with laser treatment groups (active versus inactive) were estimated. Chi-square analyses were performed to estimate for the associations between categorical demographic and clinical predictors and laser treatment groups. Fisher’s exact tests were conducted when data did not meet the assumptions of Pearson’s chi-square (cells with expected counts < 5). Independent group t tests were used to compare groups on continuous variables.
Results
Participant characteristics
At baseline, the demographic and clinical predictors of the two treatment groups were comparable, as no significant differences existed between both groups in terms of BMI, age, treatment variables, axillary lymph node procedures, and numbers of lymph nodes removed (Table 4). As a result, the randomization scheme created two relatively similar patient profiles. The mean age of both groups was between 60 and 64 years of age. Seventy-three percent of the women in the laser group were married, while only 30% of the women in the placebo group were married. Employment status was similar in both groups, with 64% employed in the laser group and 70% in the placebo group. Approximately 60% of the women in both groups had mastectomy, 90% underwent chemotherapy, and over 90% had radiation and axillary lymph node dissection. The mean number of lymph nodes removed for women in the placebo group was 16 total nodes with 5 of them positive, and for women in the laser group was 17 total nodes with 8 of them positive. Less than 10% of all participants in the study reported a family history of lymphedema.
Outcome variables: (Table 5)
Lymphedema symptoms
In comparison with the placebo group (83.3%), significantly fewer participants in the laser group (55.6%) reported more than one lymphedema symptom (p = 0.012) at 12-months post-intervention. Significantly more patients in the laser group (44.4%) reported less than two of the seven symptoms of impaired limb mobility (limb mobility in shoulder, arm, elbow, wrist and fingers, tightness and stiffness) in comparison with the placebo group (33.3%) at 12-month post-intervention (p = 0.017). There were no significant changes in symptoms of fluid accumulation (arm swelling, breast swelling, chest wall swelling, seroma, heaviness, and firmness) and pain/discomfort symptoms (pain/aching/soreness, numbness, tenderness, redness, blistering, burning, stabbing, tingling, and pain/discomfort).
Symptom distress
The laser group had statistically significant improvements in symptom distress of sadness (p = 0.005) from 73 to 11% and self-perception (p = 0.03) from 36 to 0% over time from visit 1 to visit 6. The placebo group did not show a correspondingly significant decrease in the reporting of any total number of symptoms, impaired limb mobility, symptom distress, or improvements in social functioning. There were no significant improvements in the dimensions of daily living, sleep disturbance, sexuality, and days absent from work.
Limb volume
The laser group had a significant decrease in the percentage of participants from 100 to 63% (p = 0.118) who had > 5% limb volume prior to intervention to < 5% limb volume after intervention, while the placebo group did not achieve a significant decrease (100 to 78%; p = 0.614). Yet, the changes were not statistically significant in either the laser or placebo groups.
Adherence rates
The placebo group had a significant decrease in adherence rates from 100 to 60% (p = 0.037), while the laser group maintained an adherence rate above 80% throughout the entire study (p = 0.932).
Discussion
Results from this trial showed that the addition of LLLT to CDT did help patients significantly improve their lymphedema symptom experience by decreasing the number of lymphedema symptoms and relieving impaired limb mobility symptoms. LLLT also helped to improve emotional distress from lymphedema symptoms, including sadness and self-perception. By reducing the number of lymphedema symptoms, relieving symptoms of impaired limb mobility, and improving emotional distress, LLLT played an important role in treating lymphedema, focusing on symptom management in breast cancer patients with lymphedema. Inconsistent with other studies [25, 36, 44], findings of our trial did not show statistically significant changes in limb volume in either group, although the laser group does show a decrease in limb volume of > 5% from 100 to 63%. The insignificance of limb volume in this study may be a result of the small power with a sample size n = 21.
Future research should have a larger sample size focusing on the effect of laser treatment on limb volumes. Research suggests, however, that arm volume reduction alone may not provide adequate relief of symptoms and may contribute to poorer QOL among breast cancer patients [13, 14]. Ridner found that cumulative symptom experience was found to be influenced more by the perception of arm volume than by the objective arm volume. Thus, lymphedema symptoms and symptom distress may be more sensitive indicators for evaluating the effects of lymphedema treatment [13].
Adherence to CDT during the initial intensive phase of treatment and the maintenance phase of therapy has been identified as an obstacle for long-term self-management of this condition to achieve optimal outcomes [45]. Our trial found that significantly more patients in the placebo group failed to follow up at the research visits while over 80% of the patients in the laser group maintained adherence to research follow-up visits. With a greater understanding of the impact of symptom burden reduction resulting from treatment intervention, individuals who make significant gains in symptom reduction and decreased pain from treatment intervention may be more motivated to adhere to their treatment protocol and to long-term follow-up visits in the future.
The ultimate goal of lymphedema treatment is to relieve suffering. Based on results from this study, LLLT should be considered a complementary therapy to CDT to relieve symptoms and symptom distress, in turn helping patients to achieve optimal outcomes of CDT by motivating patient adherence.
Strengths and limitations of the trial
The strengths of the study included a rigorous study design with a long-term follow-up of 12 months, while a majority of published studies utilized a shorter follow-up period of approximately 3–6 months. Outcome measures in this study included a combination of subjective and objective measurements with the ability to not only capture changes in symptoms and symptom distress but also limb volume by infrared perometer, a more rigorous measure of limb volume than tape measures used by a majority of the studies [25, 31, 32, 34,35,36, 44]. Another strength in this study was the use of a standardized fabric grid during treatment sessions to insure consistency in the location of treatment points. The grid had four different color-coded groups of holes which enabled the use of alternating laser points of application from session to session to ensure quality and safety of treatment.
There are several limitations of this study. Because the goal of the study was to examine the use of the LLL as a complementary treatment to CDT, the study protocol required continuous adherence to CDT during the treatment portion of the study and required long-term follow-up appointments. Both of these requirements presented challenges in the recruitment of subjects, as many potential participants for the study were unwilling to participate for one or both reasons. There were several enrolled subjects who were unable to complete the study. Only one subject in the laser group dropped out of the study as she complained about discomfort in the chest wall. Other participants were lost to follow-up for reasons unrelated to the study (Fig. 1). As a result of these challenges, the sample size of the study was small, so findings from this study should be considered with caution. Future research should investigate the effectiveness of LLLT as sole treatment modality for lymphedema in comparison to LLLT as the complementary therapy to CDT.
Conclusion
Results from this pilot study show significant benefit of LLLT in combination with CDT for symptom burden reduction in individuals with BCRL, but did not show additional benefit for limb volume reduction. In the future, best practice for BCRL assessment would include a combination of limb volume measurement and comprehensive symptom assessment, as this pathology has been shown to have an impact in both areas. For future studies, a larger sample size with a rigorous study design focusing on the effectiveness of LLLT as the sole treatment for the BCRL population is indicated to make more definitive conclusions.
References
Lawenda BD, Mondry TE, Johnstone PA (2009) Lymphedema: a primer on the identification and management of a chronic condition in oncologic treatment. CA Cancer J Clin 59(1):8–24
Warren AG, Brorson H, Borud LJ, Slavin SA (2007) Lymphedema: a comprehensive review. Ann Plast Surg 59(4):464–472
Rockson SG (2006) Addressing the unmet needs in lymphedema risk management. Lymphat Res Biol 4(1):42–46
Moseley ALCC, Piller NB (2007) A systematic review of common conservative therapies for arm lymphedema secondary to breast cancer treatment. Ann Oncol 18(4):639–646
Norman SA, Localio AR, Potashnik SL, Simoes Torpey HA, Kallan MJ, Weber AL et al (2009) Lymphedema in breast cancer survivors: incidence, degree, time course, treatment, and symptoms. J Clin Oncol 27(3):390–397
Fu MR, Deng J, Armer JM. Putting evidence into practice: cancer-related lymphedema. Clin J Oncol Nurs 2014;18 Suppl:68–79
Armer JM, Radina ME, Porock D, Culbertson SD (2003) Predicting breast cancer-related lymphedema using self-reported symptoms. Nurs Res 52(6):370–379
Fu MR, Rosedale M (2009) Breast cancer survivors’ experiences of lymphedema-related symptoms. J Pain Symptom Manag 38(6):849–859
Kosir MA, Rymal C, Koppolu P, Hryniuk L, Darga L, Du W et al (2001) Surgical outcomes after breast cancer surgery: measuring acute lymphedema. J Surg Res 95(2):147–151
Ridner SH, Deng J, Fu MR, Radina E, Thiadens SR, Weiss J et al (2012) Symptom burden and infection occurrence among individuals with extremity lymphedema. Lymphology 45(3):113–123
Fu MR, Axelrod D, Haber J (2008) Breast-cancer-related lymphedema: information, symptoms, and risk-reduction behaviors. J Nurs Scholarsh 40(4):341–348
Passik SD, McDonald MV (1998) Psychosocial aspects of upper extremity lymphedema in women treated for breast carcinoma. Cancer 83(12 Suppl American):2817–2820
Ridner SH (2005) Quality of life and a symptom cluster associated with breast cancer treatment-related lymphedema. Support Care Cancer 13(11):904–911
Ahmed RL, Prizment A, Lazovich D, Schmitz KH, Folsom AR (2008) Lymphedema and quality of life in breast cancer survivors: the Iowa women’s health study. J Clin Oncol 26(35):5689–5696
Shaitelman SF, Cromwell KD, Rasmussen JC, Stout NL, Armer JM, Lasinski BB et al (2015) Recent progress in the treatment and prevention of cancer-related lymphedema. CA Cancer J Clin 65(1):55–81
Lasinski BB, McKillip Thrift K, Squire D, Austin MK, Smith KM, Wanchai A et al (2012) A systematic review of the evidence for complete decongestive therapy in the treatment of lymphedema from 2004 to 2011. PM R 4(8):580–601
Ridner SH, Fu MR, Wanchai A, Stewart BR, Armer JM, Cormier JN (2012) Self-management of lymphedema: a systematic review of the literature from 2004 to 2011. Nurs Res 61(4):291–299
McLaughlin SA, DeSnyder SM, Klimberg S, Alatriste M, Boccardo F, Smith ML et al (2017) Considerations for clinicians in the diagnosis, prevention, and treatment of breast cancer-related lymphedema, recommendations from an expert panel: part 2: preventive and therapeutic options. Ann Surg Oncol 24(10):2827–2835
Oremus M WK, Dayes I et al. Diagnosis and treatment of secondary lymphedema technology assessment report project ID: LYMT0908 May 28, 2010. McMaster University Evidence-based Practice Center. http://www.cms.gov/Medicare/Coverage/DeterminationProcess/downloads/id66aTA.pdf. Accessed 3 February 2019
Mayrovitz H (2009) The standard of care for lymphedema: current concepts and physiological considerations. Lymphat Res Biol 7(2):101–108
Finnane A, Janda M, Hayes SC (2015) Review of the evidence of lymphedema treatment effect. Am J Phys Med Rehabil 94(6):483–498
McNeely ML, Peddle CJ, Yurick JL, Dayes IS, Mackey JR (2011) Conservative and dietary interventions for cancer-related lymphedema: a systematic review and meta-analysis. Cancer. 117(6):1136–1148
Paskett ED, Dean JA, Oliveri JM, Harrop JP (2012) Cancer-related lymphedema risk factors, diagnosis, treatment, and impact: a review. J Clin Oncol 30(30):3726–3733
Smoot B, Chiavola-Larson L, Lee J, Manibusan H, Allen DD (2015) Effect of low-level laser therapy on pain and swelling in women with breast cancer-related lymphedema: a systematic review and meta-analysis. J Cancer Surviv 9(2):287–304
Ridner SH, Poage-Hooper E, Kanar C, Doersam JK, Bond SM, Dietrich MS (2013) A pilot randomized trial evaluating low-level laser therapy as an alternative treatment to manual lymphatic drainage for breast cancer-related lymphedema. Oncol Nurs Forum 40(4):383–393
E Lima MT, E Lima JG, de Andrade MF, Bergmann A (2014) Low-level laser therapy in secondary lymphedema after breast cancer: systematic review. Lasers Med Sci 29(3):1289–1295
Omar MT, Shaheen AA, Zafar H (2012) A systematic review of the effect of low-level laser therapy in the management of breast cancer-related lymphedema. Support Care Cancer 20(11):2977–2984
Wang X, Tian F, Reddy DD et al (2017) Up-regulation of cerebral cytochrome-c-oxidase and hemodynamics by transcranial infrared laser stimulation: a broadband near-infrared spectroscopy study. J Cerebr Blood F Met 37(12):3789–3802
Liebert AD, Bicknell BT, Adams RD (2014) Protein conformational modulation by photons: a mechanism for laser treatment effects. Med Hypotheses 82(3):275–281
Jang DH, Song DH, Chang EJ, Jeon JY (2016) Anti-inflammatory and lymphangiogenetic effects of low-level laser therapy on lymphedema in an experimental mouse tail model. Lasers Med Sci 31(2):289–296
Kaviana A, Fateh M, Yousefi-Nooraie R, Alinagi-Zadeh M, Ataie-Fashtami L (2006) Low-level laser therapy in management of postmastectomy lymphedema. Lasers Med Sci 21(2):90–94
Piller NB, Thelander A (1998) Treatment of chronic postmastectomy lymphedema with low level laser therapy: a 2.5 year follow-up. Lymphology 31(2):74–86
Carati CJ, Anderson SN, Gannon BJ, Piller NB (2003) Treatment of postmastectomy lymphedema with low-level laser therapy: a double blind, placebo-controlled trial. Cancer 98(6):1114–1122
Dirican A, Andacoglu O, Johnson R, McGuire K, Mager L, Soran A (2011) The short-term effects of low-level laser therapy in the management of breast-cancer-related lymphedema. Support Care Cancer 19(5):685–690
Kozanoglu E, Basaran S, Paydas S, Sarpel T (2009) Efficacy of pneumatic compression and low-level laser therapy in the treatment of postmastectomy lymphoedema: a randomized controlled trial. Clin Rehabil 23(2):117–124
Omar MTA, Ebid AA, El Morsy AM (2011) Treatment of post-mastectomy lymphedema with laser therapy: double blind placebo control randomized study. J Surg Res 165(1):82–90
Available from: https://www.accessdata.fda.gov/cdrh_docs/pdf3/K030295.pdf. Accessed 3 February 2019
Fu MR, Axelrod D, Guth AA, Cartwright F, Qiu Z, Goldberg JD et al (2014) Proactive approach to lymphedema risk reduction: a prospective study. Ann Surg Oncol 21(11):3481–3489
Fu MR, Axelrod D, Guth AA, Fletcher J, Qiu JM, Scagliola J et al (2015) Patterns of obesity and lymph fluid level during the first year of breast cancer treatment: a prospective study. J Pers Med 5(3):326–340
Fu MR, Axelrod D, Cleland CM, Qiu Z, Guth AA, Kleinman R et al (2015) Symptom report in detecting breast cancer-related lymphedema. Breast Cancer (Dove Med Press) 7:345–352
Fu MR, Axelrod D, Guth AA, Rampertaap K, El-Shammaa N, Hiotis K et al (2016) mHealth self-care interventions: managing symptoms following breast cancer treatment. Mhealth. 2
Shi S, Lu Q, Fu MR, Ouyang Q, Liu C, Lv J et al (2016) Psychometric properties of the breast cancer and lymphedema symptom experience index: the Chinese version. Eur J Oncol Nurs 20:10–16
Fu MR, Conley YP, Axelrod D, Guth AA, Yu G, Fletcher J et al (2016) Precision assessment of heterogeneity of lymphedema phenotype, genotypes and risk prediction. Breast 29:231–240
Maiya AG, Olivia E, Dibya A (2008) Effect of low energy laser therapy in the management of post-mastectomy lymphedema. Physio Singapore 11(1):2–5
Mondry TE, Riffenburgh RH, Johnstone PA (2004) Prospective trial of complete decongestive therapy for upper extremity lymphedema after breast cancer therapy. Cancer J 10(1):42–48 discussion 17-9
Acknowledgments
We thank RianCorp for loaning us the active and inactive low-level laser devices.
Author information
Authors and Affiliations
Contributions
(1) Conception and design: Mei R. Fu, Ting-Ting Kuo, Laurie Kilmartin, Tara Denham.
(2) Administrative support: Tara Denham.
(3) Provision of study material or patients: Deborah Axelrod, Amber A. Guth.
(4) Data collection and assembly: Mei R. Fu, Laurie Kilmartin, Ting-Ting Kuo, Aaron Beattie.
(5) Data analysis and interpretation: Mei R. Fu, Gary Yu, Laurie Kilmartin, Tara Denham.
(6) Manuscript writing: All authors.
(7) Final approval of manuscript: All authors.
Corresponding author
Ethics declarations
Disclosure
The design and contents of the study are solely the responsibility of the authors and do not necessarily represent the views of RianCorp. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
List of products used: RianCorp LTU 904 low-level laser.
Conflict of interest
The authors declare no conflict of interest except the loan of the active and inactive low-level laser devices.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kilmartin, L., Denham, T., Fu, M.R. et al. Complementary low-level laser therapy for breast cancer-related lymphedema: a pilot, double-blind, randomized, placebo-controlled study. Lasers Med Sci 35, 95–105 (2020). https://doi.org/10.1007/s10103-019-02798-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10103-019-02798-1