
Abstract
Periodontal disease is a chronic inflammatory disease that is commonly treated with surgical and nonsurgical techniques. However, both approaches have limitations. Low-level laser therapy (LLLT) has been widely applied in reducing inflammatory reactions, and research indicates that LLLT induces an anti-inflammatory effect that may enhance periodontal disease therapy. The purpose of this study was to investigate the anti-inflammatory effect of LLLT on human periodontal ligament cells (hPDLCs) in an inflammatory environment and aimed to determine the possible mechanism of action. Cells were cultured and treated with or without lipopolysaccharide (LPS) from Porphryromonas gingivalis or Escherichia coli, followed by irradiation with a gallium-aluminum-arsenide (GaAlAs) laser (660 nm) at an energy density of 8 J/cm2. Quantitative real-time polymerase chain reactions were used to assess the expression of pro-inflammatory genes, including tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, IL-6, and IL-8. The dual-luciferase reporter assay was used to examine nuclear factor-κB (NF-κB) transcriptional activity. An enzyme-linked immunosorbent assay was used to monitor the concentration of intracellular cyclic adenosine monophosphate (cAMP). Both LPS treatments significantly induced the mRNA expression of pro-inflammatory cytokines. However, LLLT inhibited the LPS-induced pro-inflammatory cytokine expression and elevated intracellular levels of cAMP. The LLLT inhibitory effect may function by downregulating NF-κB transcriptional activity and by increasing the intracellular levels of cAMP. LLLT might inhibit LPS-induced inflammation in hPDLCs through cAMP/NF-κB regulation. These results should be further studied to improve periodontal therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Periodontal disease is a common and complex inflammatory disease that results in progressive destruction of the tooth-supporting soft and hard tissues of the periodontium [1]. The resulting damage affects the alveolar bone as well as the periodontal ligament (PDL). Although surgical therapies such as open flap surgery and osseous surgery and nonsurgical therapies such as standard oral hygiene procedure and sustained local delivery antimicrobial agents are helpful in controlling this disease, both treatment options have limitations. Patients with deep pockets or furcation involvement did not respond well to nonsurgical therapy, and surgical therapy was more invasive and risky when treating patients with systemic problems [2, 3].
It has been reported that one of the functions of PDL cells is to respond to environmental stresses, such as inflammatory cytokines and pathogens [4, 5]. Sun et al. reported that lipopolysaccharide (LPS) exposure induces the expression of cytokines in PDL cells, including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, and IL-8 [4]. Inflammatory cells as well as PDL cells regulate the periodontitis process [4,5,6], although the exact roles of different cell types require further clarification.
LPS is a macromolecule with a lipid element (lipid A) and a polysaccharide element [7]. LPS is located on the exterior membrane of gram-negative bacteria and stimulates powerful immune responses in animals. Indeed, the immune systems of animals have evolved to recognize LPS via toll-like receptors (TLRs) [8]. TLRs are a class of transmembrane proteins that contain numerous leucine-rich repeats and are composed of globular intracellular regions and extracellular domains [9]. Eleven different TLRs in humans have been identified [10]. Among them, TLR2 and 4 function as the principal innate sensors for the virulent factors of periodontopathic bacteria [11, 12]. LPS binding to TLR 2 and 4 triggers activation of the nuclear factor kappa B (NF-κB) signaling cascade, inducing the secretion of pro-inflammatory cytokines such as TNF-α and IL-1β [13]. Alterations to the number, position, and length of the acyl groups or monosaccharide groups in lipid A will result in alterations in the biologic effects [14]. Based on these mechanisms, LPS from different bacterial species may trigger different types of immune responses.
In the past few decades, low-level laser therapy (LLLT) has been widely employed in the treatment of wound healing, musculoskeletal pain, and chronic and acute inflammation [15, 16]. Studies have identified the benefits of using LLLT [17, 18]. For example, Bortone et al. reported that LLLT induced an anti-inflammatory effect through the regulation of pro-inflammatory mRNAs [17]. Obradovic et al. reported that low-level lasers have shown efficacy in reducing inflammation of periodontitis in patients with diabetes mellitus [18]. However, the precise mechanism by which LLLT regulates periodontal inflammation remains unknown.
Cyclic adenosine monophosphate (cAMP) functions as an important second messenger in many cell physiological processes, such as cell differentiation, proliferation, apoptosis, and inflammation [19]. cAMP is formed by the activation of adenylyl cyclase and is converted from adenosine triphosphate [19]. Also, cAMP was recently implicated in pro-inflammatory processes as it was shown to not only serve as a modulator of immune function but also as a regulator in response to LLLT signaling [20]. However, the potential relationship between cAMP and LLLT in affecting the LPS-induced inflammatory response in human periodontal ligament cells (hPDLCs) has not been investigated.
In this study, we investigated the anti-inflammatory effects of LLLT on hPDLCs that were exposed to LPS from Porphryromonas gingivalis (P. gingivalis) or Escherichia coli (E. coli) and aimed to determine the possible mechanism of action.
Materials and methods
Cell culture
The procedure for hPDL cell preparation was according to a modification of the method reported by D’Errico et al. [21]. Briefly, hPDL tissues were collected from the middle third of periodontally healthy and non-carious third molar roots extracted from healthy volunteers (20 to 40 years of age) during the course of orthodontic treatment. Periodontal ligament tissue was scraped from third molars, enzymatically digested by 2 mg/mL collagenase and 0.25% trypsin for 1 h at 37 °C. The cells were harvested by centrifugation for 10 min at 500×g. The supernatant was carefully aspirated, and cells were washed twice with Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum. The cells were expanded with DMEM supplemented with 10% fetal bovine serum, 100 mg/mL penicillin-G, 100 mg/mL kanamycin sulfate, and 0.3 mg/mL amphotericin B. The cultures were kept at 37 °C in a humidified incubator in the presence of 95% air and 5% CO2. The medium was changed every other day. The cells were used at passages 3 to 9.
Chemical inhibitor and reagent treatments
LPS from E. coli and P. gingivalis was dissolved in 1× phosphate-buffered saline at a final concentration of 10 mg/mL and stored at 4 °C. The hPDLCs were treated with different concentrations of LPS (0, 10, 20, 50, and 100 μg/mL) from P. gingivalis and E. coli freshly diluted in culture medium. The adenylyl cyclase inhibitor SQ22536 (Sigma-Aldrich, St. Louis, MO, USA) was used to block the activity of adenylyl cyclase. SQ22536 was dissolved in dimethyl sulfoxide (DMSO), and the cells were treated with a dose of 100 μmol/L. Forskolin (50 μmol/L, Sigma-Aldrich, St. Louis, MO, USA), an adenylyl cyclase activator, was dissolved in DMSO.
Low-level laser therapy
A gallium-aluminum-arsenide red laser with flat top beam profile (wavelength 660 nm diode laser, Transverse Industries Co., Ltd., Taipei, Taiwan) was used as the laser source. The output of the laser device was 70 mW. The distance between the bottom of the culture plate and the laser source was 3 cm, and the irradiated area was 3.8 cm2. The cells were irradiated once for 528 s in a continuous mode and received 8 J/cm2 of laser energy density in total. All irradiation experiments were performed on a clean bench at room temperature. The control groups were processed under the same conditions, except without laser irradiation.
Cell viability
An MTT assay (3-[4, 5- dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide) was used to determine cell viability. The hPDLCs were seeded onto a 96-well plate. After exposure to LPS from P. gingivalis and E. coli for 24 h, the cells were incubated with 5 mg/mL MTT for 4 h. The reactions were measured using an enzyme-linked immunosorbent assay (ELISA) reader at 595 nm.
Lactate dehydrogenase leakage
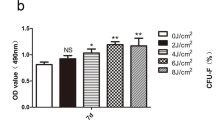
Lactate dehydrogenase (LDH) leakage was measured to quantify cytotoxicity with a Cytotoxicity Detection kit. The hPDLCs were seeded onto a 96-well plate and cultured with different concentrations of LPS from P. gingivalis and E. coli for 24 h. The LDH leakage was calculated according to the manufacturer’s guidelines. The absorbance was measured with an ELISA reader at 490 nm.
Real-time reverse transcription-polymerase chain reaction (RT-PCR)
The total RNA from hPDLCs was extracted using TRIzol (Thermo Fisher Scientific, Waltham, MA, USA). cDNA reverse transcription was conducted with an RT system containing Moloney Murine Leukemia Virus reverse transcriptase. Quantitative real-time PCR was performed in a Bio-Rad CFX Connect real-time PCR detecting system, and the reactions were carried out in a mixture containing cDNA, primers for each gene, and iQ™ Syber-Green Supermix (Bio-Rad, Hercules, CA, USA). The primer pair sequences for the following human genes are listed in Table 1. The relative mRNA expression levels were analyzed with the comparative Ct method, where the amount of target is normalized to the housekeeping gene GAPDH.
Intracellular cyclic adenosine monophosphate levels
ELISA kits for the detection of cAMP were obtained from Enzo (Farmingdale, NY, USA). All of the procedures were based on the manufacturer’s instructions, and the optical density was measured at a wavelength of 405 nm.
Dual-luciferase assay
We cultured 2 × 104 hPDLCs in a 48-well plate and co-transfected the cells with the pGL 4.32 [luc2P/NF-κB-RE/Hygro] vector (250 ng) and pTK-Renilla vector (50 ng) using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA). The transfection procedure was performed according to the manufacturer’s instructions. Twenty-four hours after transfection, the cells were treated with LPS, LLLT, and SQ22536 for an additional 6 h. The luciferase activity was measured using a dual-luciferase reporter assay system (Promega, Madison, WI, USA) with an Anthos Lucy3 microplate luminometer (AnthosLabtec Instruments, Austria).
Statistical analysis
All the data were collected from three to four independent experiments. Data were analyzed with the statistical software SPSS/Win version 17.0 and expressed as the mean ± standard deviation. For statistical analysis, normality was first tested using the Shapiro-Wilk test. For normally distributed data, one-way analysis of variance was used to test for statistical differences followed by the post hoc Tukey’s test. If the data failed the normality test, the nonparametric Kruskall-Wallis one-way analysis of variance was used to test for statistical differences with Dunn’s multiple comparison test. A p value of 0.05 was considered statistically significant.
Results
The viability and cytotoxicty of LPS-challenged hPDLCs
To evaluate the influence of LPS from P. gingivalis and E. coli on the viability of hPDLCs, an MTT test was conducted. The MTT assay showed that the viability of hPDLCs was not reduced following treatment with P. gingivalis or E. coli LPS at 24 h (Fig. 1a, b). The cytotoxic effects of LPS on hPDLCs were also measured with the LDH leakage assay. After normalizing to the control group, the activities of LDH leakage showed no differences between the control and LPS-treated groups at 24 h (Fig. 1c, d). These results indicated that LPS was not cytotoxic and did not reduce the viability of hPDLCs in the short time intervals. All subsequent experiments were analyzed in 24-h increments.

LPS did not induce cytotoxic effects nor reduce the viability of hPDLCs. The hPDLCs were treated with LPS from P. gingivalis or E. coli doses of 0 (control), 10, 20, 50, or 100 μg/mL. MTT assays (a, b) and LDH leakage analysis (c, d) were used to evaluate cell viability and cytotoxicity. The data are shown as the mean ± SD. N = 3 (N numbers of independent experiments). There were no significant differences between the groups
The expression patterns of inflammatory cytokines in LPS-challenged hPDLCs
The hPDLCs were treated with different concentrations of LPS from P. gingivalis or E. coli for 24 h. Then, q-PCR was conducted to assess the gene expression of pro-inflammatory cytokines including IL-1β, TNF-α, IL-6, and IL-8. The results showed that LPS from P. gingivalis or E. coli enhanced the mRNA expression of TNF-α, IL-1β, IL-6, and IL-8 in a dose-dependent manner with increasing LPS concentrations. LPS from P. gingivalis induced a 2- to 4-fold elevation of mRNA expression at a lower dose (20 μg/mL), while LPS from E. coli had the same effect at a higher dose (50 μg/mL) (Fig. 2). At doses higher than 50 μg/mL, the expression levels of most pro-inflammatory cytokine genes were observed to plateau (Fig. 2a, c–f).

hPDLCs stimulated by LPS expressed inflammatory genes. LPS from P. gingivalis or E. coli increased the mRNA expression of IL-1β, TNF-α, IL-6, and IL-8. a–d Cells treated with LPS from P.gingivalis. e–h Cells treated with LPS from E. coli. The data are shown as the mean ± SD. N = 4 (N numbers of independent experiments). The following statistical levels were applied: *p < 0.05 compared with the 0 μg/mL group (control); †p < 0.05 compared with the 10 μg/mL group; ‡p < 0.05 compared with the 20 μg/mL group
The anti-inflammation effect of LLLT on hPDLCs
The hPDLCs were treated with or without LPS from P. gingivalis (20 μg/mL) or E. coli (50 μg/mL), which was immediately followed by laser irradiation at an energy density of 8 J/cm2 to determine the effect of LLLT on the LPS-induced inflammatory response. Twenty-four hours following the exposure of hPDLCs to LPS from P. gingivalis or E. coli, there was an elevated level of inflammatory cytokine mRNA expression in the non-irradiated group. The group treated with LPS and LLLT showed obvious decreases in cytokine mRNAs compared to the LPS-only group (Fig. 3).

A cAMP inhibitor (SQ22536) hindered the anti-inflammatory effect of LLLT on inflammation induced by LPS of P. gingivalis and E. coli. Real-time RT-PCR was performed to measure the mRNA levels of a IL-1β, b TNF-α, c IL-6, and d IL-8. The results were analyzed with the 2−ΔCT method based on the control. The data are shown as the mean ± SD. N = 4 (N numbers of independent experiments). The following statistical levels were applied: *p < 0.05 compared with the control group and †p < 0.05
To elucidate whether LLLT inhibited inflammatory cytokine mRNA expression by regulating cAMP, we treated hPDLCs with SQ22536, a pharmacological inhibitor of cAMP, and then treated with LPS and LLLT. In this group, the level of pro-inflammatory cytokine mRNA expression was not reduced by LLLT (Fig. 3).
Next, an ELISA was conducted to examine the cAMP levels associated with LLLT in hPDLCs. When hPDLCs were treated with the adenylyl cyclase activator forskolin or irradiated with the low-level laser, the level of cAMP increased and showed a significant difference compared to the control group. However, when hPDLCs were treated with SQ22536 and irradiated with the low-level laser, the level of cAMP was not elevated and showed a significantly lower level when compared to the LLLT or forskolin-only groups (Fig. 4). These results indicated that LLLT may reduce inflammation through the regulation of cAMP. We further investigated whether LLLT affected the transcriptional activity of NF-κB, a crucial inflammatory transcription factor, using luciferase reporter assays. When hPDLCs were treated with LPS from P. gingivalis (20 μg/mL) or E. coli (50 μg/mL), the level of NF-κB was increased and showed a significant difference compared to the control group (Fig. 5). When hPDLCs were treated with LPS and irradiated with the low-level laser, the level of NF-κB showed no significant difference compared to the control group. With the addition of SQ22536, the irradiated LPS-stimulated hPDLCs showed a similar NF-κB level as the LPS-only group and a major difference when compared to the control and laser-irradiated LPS-stimulated groups (Fig. 5). These results suggested that LLLT regulated NF-κB transcriptional activity by affecting the cAMP level.

LLLT increased the intracellular cAMP levels. Intracellular cAMP levels were measured by ELISA in hPDLCs treated with LLLT, cAMP inhibitor (SQ22536), or forskolin. The data are shown as the mean ± SD. N = 4 (N numbers of independent experiments). The following statistical levels were applied: *p < 0.05 compared with the control group; †p < 0.05 compared with the forskolin-only group; ‡p < 0.05 compared with the LLLT-only group

LLLT reduced NF-κB transcriptional activity. The NF-κB luciferase activity was elevated by LPS and reduced by LLLT. With the addition of the cAMP inhibitor (SQ22536), the NF-κB activity in LPS-stimulated hPDLCs was not reduced by LLLT: a LPS from P. gingivalis; b LPS from E. coli. The data are shown as the mean ± SD. N = 4 (N numbers of independent experiments). The following statistical levels were applied: *p < 0.05 compared with control group; †p < 0.05
Discussion
The hPDLCs are human fibroblasts with multifunctional tissue that provide physical, formative and remodeling, nutritional, and sensory functions [22]. In the present study, the hPDLCs were treated with different concentrations of LPS from P. gingivalis or E. coli. Our data showed that LPS from both P. gingivalis and E. coli induced pro-inflammatory cytokine (IL-1β, TNF-α, IL-6, IL-8) mRNA expressions in a dose-dependent manner. However, P. gingivalis-treated cells responded in a dose-dependent manner at lower doses when compared to E. coli-treated cells (Fig. 2). The different levels of induced inflammation from the two sources may be attributed to molecular differences in the lipid components. The lipid A from P. gingivalis LPS is a monophosphate type lacking a phosphate group in the 4′ position that does not contain a tetradecanoic acid, but rather long-chain fatty acids made up of only acyloxyl groups [23]. It is believed that LPS from P. gingivalis is recognized mainly by TLR2, while LPS from E. coli is recognized mainly by TLR4 [23,24,25,26]. Uehara et al. also reported that human gingival fibroblasts showed more robust mRNA expression of pro-inflammatory cytokines following TLR2 ligation (Mycoplasma-type diacyl lipopeptide) compared to TLR4 ligation (E. coli-type lipid A) [27]. These results indicate that different sources of LPS might induce inflammation through different mechanisms.
In recent decades, many studies have shown that LLLT reduces inflammatory reactions both in vitro and in vivo. Correa et al. induced periodontitis in mice with LPS and found that LLLT (GaAs laser, 904 nm) diminished inflammatory cell migration in a dose-dependent manner, with an energy dose of 3 J/cm2 identified as the most effective dose [28]. Pires et al. reported a model of collagenase-induced tendinitis and demonstrated that LLLT (780 nm), at an energy dose of 7.7 J/cm2, suppressed the expression of IL-6 [29]. Boschi et al. reported that LLLT (InGaAlP laser, 660 nm) significantly reduced the expression of IL-6 and TNF-α [30]. According to previous studies conducted in our lab [31], LLLT (GaAlAs laser, 660 nm) showed the most effective suppression of inflammation at the optimal dose of 8 J/cm2, which was used in this study. In the current study, we noted that LPS-challenged hPDLCs showed similar results to the aforementioned studies, suggesting that LLLT significantly suppressed the mRNA expression of pro-inflammatory cytokines (IL-1β, TNF-α, IL-6, IL-8), leading us to conclude that LLLT has an anti-inflammatory effect in hPDLCs (Fig. 3). In addition, we noticed that the anti-inflammatory effect of LLLT was neutralized when the cAMP inhibitor SQ22536 was used (Fig. 3). We also observed that the level of cAMP from hPDLCs was elevated by both forskolin (cAMP promoter) and LLLT but decreased when SQ22536 (cAMP inhibitor) was added (Fig. 4), consistent with previous study that the level of cAMP increased by approximately 3- to 4-fold following LLLT treatment of human adipose-derived stem cells [31]. This finding suggests that LLLT can act to stimulate the level of cAMP. Other studies have shown that the elevation of intracellular cAMP levels inhibited the transcriptional activity of NF-κB, which is a crucial transcription factor in the regulation of inflammation [32, 33]. Possible mechanisms that regulate of NF-κB activity include the ability of cAMP to manage IκB degradation and IKK activity as well as to change the composition of NF-κB dimers and thereby block transcription [34]. Indeed, we observed that LLLT significantly inhibited the transcriptional activity of NF-κB in our previous study of LPS-stimulated human adipose-derived stem cells [31]. In the present study, we treated LPS-stimulated hPDLCs with LLLT and the adenylyl cyclase inhibitor SQ22536 and observed that the inhibitory effect of LLLT on NF-κB transcriptional activity was significantly reduced (Fig. 5). Aimbire et al. [35] also reported results similar to ours, showing that a low-level laser (660 nm) at an energy dose of 7.5 J/cm2 inhibited NF-κB transcriptional activity and further reduced apoptotic gene expression.
Exploring LLLT’s role in regulating the induction of pro-inflammatory cytokine in periodontal pathology is important as it may lead to novel therapeutic approaches for periodontitis. This study has demonstrated that LLLT inhibits inflammation, induced by LPS from E. coli and P. gingivalis, through the cAMP/NF-ĸB signaling pathway in hPDLCs. Future research into the detailed regulation of LLLT on cAMP may be of great value in improving periodontal therapy.
References
Yoshinaka K, Shoji N, Nishioka T, Sugawara Y, Hoshino T, Sugawara S, Sasano T (2014) Increased interleukin-18 in the gingival tissues evokes chronic periodontitis after bacterial infection. Tohoku J Exp Med 232(3):215–222
Drisko CH (2001) Nonsurgical periodontal therapy. Periodontol 2000(25):77–88
Wang HL, Greenwell H (2001) Surgical periodontal therapy. Periodontol 25:89–99
Sun Y, Shu R, Li CL, Zhang MZ (2010) Gram-negative periodontal bacteria induce the activation of Toll-like receptors 2 and 4, and cytokine production in human periodontal ligament cells. J Periodontol 81(10):1488–1496
Jonsson D, Nebel D, Bratthall G, Nilsson BO (2011) The human periodontal ligament cell: a fibroblast-like cell acting as an immune cell. J Periodontal Res 46(2):153–157
Yamaji Y, Kubota T, Sasaguri K, Sato S, Suzuki Y, Kumada H, Umemoto T (1995) Inflammatory cytokine gene expression in human periodontal ligament fibroblasts stimulated with bacterial lipopolysaccharides. Infect Immun 63(9):3576–3581
Simpson BW, May JM, Sherman DJ, Kahne D, Ruiz N (2015) Lipopolysaccharide transport to the cell surface: biosynthesis and extraction from the inner membrane. Philos Trans R Soc Lond B Biol Sci 370(1679)
Carpenter S, O'Neill LA (2007) How important are Toll-like receptors for antimicrobial responses? Cell Microbiol 9(8):1891–1901
Doyle SL, O'Neill LA (2006) Toll-like receptors: from the discovery of NFkappaB to new insights into transcriptional regulations in innate immunity. Biochem Pharmacol 72(9):1102–1113
Essakalli M, Atouf O, Bennani N, Benseffaj N, Ouadghiri S, Brick C (2009) Toll-like receptors. Pathol Biol (Paris) 57(5):430–438
Tateishi F, Hasegawa-Nakamura K, Nakamura T, Oogai Y, Komatsuzawa H, Kawamata K, Douchi T, Hatae M, Noguchi K (2012) Detection of Fusobacterium nucleatum in chorionic tissues of high-risk pregnant women. J Clin Periodontol 39(5):417–424
Wara-aswapati N, Chayasadom A, Surarit R, Pitiphat W, Boch JA, Nagasawa T, Ishikawa I, Izumi Y (2013) Induction of toll-like receptor expression by Porphyromonas gingivalis. J Periodontol 84(7):1010–1018
O’Neill LA, Bowie AG (2007) The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 7(5):353–364
Schromm AB, Brandenburg K, Loppnow H, Moran AP, Koch MH, Rietschel ET, Seydel U (2000) Biological activities of lipopolysaccharides are determined by the shape of their lipid A portion. Eur J Biochem 267(7):2008–2013
Farivar S, Malekshahabi T, Shiari R (2014) Biological effects of low level laser therapy. J Lasers Med Sci 5(2):58–62
Arany PR (2016) Craniofacial wound healing with photobiomodulation therapy: new insights and current challenges. J Dent Res 95(9):977–984
Bortone F, Santos HA, Albertini R, Pesquero JB, Costa MS, Silva JA Jr (2008) Low level laser therapy modulates kinin receptors mRNA expression in the subplantar muscle of rat paw subjected to carrageenan-induced inflammation. Int Immunopharmacol 8(2):206–210
Obradovic R, Kesic L, Mihailovic D, Antic S, Jovanovic G, Petrovic A, Pesevska S (2013) A histological evaluation of a low-level laser therapy as an adjunct to periodontal therapy in patients with diabetes mellitus. Lasers Med Sci 28(1):19–24
Gether U (2000) Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr Rev 21(1):90–113
de Lima FM, Moreira LM, Villaverde AB, Albertini R, Castro-Faria-Neto HC, Aimbire F (2011) Low-level laser therapy (LLLT) acts as cAMP-elevating agent in acute respiratory distress syndrome. Lasers Med Sci 26(3):389–400
D’Errico JA, Ouyang H, Berry JE, MacNeil RL, Strayhorn C, Imperiale MJ, Harris NL, Goldberg H, Somerman MJ (1999) Immortalized cementoblasts and periodontal ligament cells in culture. Bone 25(1):39–47
Roberts WE, Chamberlain JG (1978) Scanning electron microscopy of the cellular elements of rat periodontal ligament. Arch Oral Biol 23(7):587–589
Sun Y, Li H, Sun MJ, Zheng YY, Gong DJ, Xu Y (2014) Endotoxin tolerance induced by lipopolysaccharides derived from Porphyromonas gingivalis and Escherichia coli: alternations in Toll-like receptor 2 and 4 signaling pathway. Inflammation 37(1):268–276
Kikkert R, Laine ML, Aarden LA, van Winkelhoff AJ (2007) Activation of toll-like receptors 2 and 4 by gram-negative periodontal bacteria. Oral Microbiol Immunol 22(3):145–151
Manicassamy S, Pulendran B (2009) Modulation of adaptive immunity with Toll-like receptors. Semin Immunol 21(4):185–193
Souza PP, Palmqvist P, Lundgren I, Lie A, Costa-Neto CM, Lundberg P, Lerner UH (2010) Stimulation of IL-6 cytokines in fibroblasts by toll-like receptors 2. J Dent Res 89(8):802–807
Uehara A, Takada H (2007) Functional TLRs and NODs in human gingival fibroblasts. J Dent Res 86(3):249–254
Correa F, Lopes Martins RA, Correa JC, Iversen VV, Joenson J, Bjordal JM (2007) Low-level laser therapy (GaAs lambda = 904 nm) reduces inflammatory cell migration in mice with lipopolysaccharide-induced peritonitis. Photomed Laser Surg 25(4):245–249
Pires D, Xavier M, Araujo T, Silva JA Jr, Aimbire F, Albertini R (2011) Low-level laser therapy (LLLT; 780 nm) acts differently on mRNA expression of anti- and pro-inflammatory mediators in an experimental model of collagenase-induced tendinitis in rat. Lasers Med Sci 26(1):85–94
Boschi ES, Leite CE, Saciura VC, Caberlon E, Lunardelli A, Bitencourt S, Melo DA, Oliveira JR (2008) Anti-inflammatory effects of low-level laser therapy (660 nm) in the early phase in carrageenan-induced pleurisy in rat. Lasers Surg Med 40(7):500–508
Wu JY, Chen CH, Wang CZ, Ho ML, Yeh ML, Wang YH (2013) Low-power laser irradiation suppresses inflammatory response of human adipose-derived stem cells by modulating intracellular cyclic AMP level and NF-kappaB activity. PLoS One 8(1):e54067
Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S (1997) The transcriptional activity of NF-kappaB is regulated by the IkappaB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell 89(3):413–424
Parry GC, Mackman N (1997) Role of cyclic AMP response element-binding protein in cyclic AMP inhibition of NF-kappaB-mediated transcription. J Immunol 159(11):5450–5456
Gerlo S, Kooijman R, Beck IM, Kolmus K, Spooren A, Haegeman G (2011) Cyclic AMP: a selective modulator of NF-kappaB action. Cell Mol Life Sci 68(23):3823–3841
Aimbire F, Santos FV, Albertini R, Castro-Faria-Neto HC, Mittmann J, Pacheco-Soares C (2008) Low-level laser therapy decreases levels of lung neutrophils anti-apoptotic factors by a NF-kappaB dependent mechanism. Int Immunopharmacol 8(4):603–605
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
This study was independently reviewed and approved by the human subjects ethics board of Kaohsiung Medical University and was conducted in accordance with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
Funding
This study was funded by the Ministry of Science and Technology of Taiwan (MOST-104-2314-B-037-059-) and Kaohsiung Medical University Aim for the Top Universities Grants (KMU-TP103B08 and KMU-TP104B11).
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lee, JH., Chiang, MH., Chen, PH. et al. Anti-inflammatory effects of low-level laser therapy on human periodontal ligament cells: in vitro study. Lasers Med Sci 33, 469–477 (2018). https://doi.org/10.1007/s10103-017-2376-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10103-017-2376-6