
Abstract
The aim of our study was to characterize the epidemiological situation concerning nosocomial vancomycin-resistant Enterococcus faecalis of VanA-phenotype (VREfs-VanA) in Poland by investigating their clonal relationships and the vanA-associated mobilome. One-hundred twenty-five clinical isolates of VREfs-VanA collected between 2004 and 2016 were studied by phenotypic assays, multilocus sequence typing (MLST), pulsed-field gel electrophoresis (PFGE), PCR detection of plasmid-specific genes, and Tn1546 structure and localization mapping. Selected isolates were subjected to PFGE-S1, Southern hybridization, genomic sequencing and conjugation experiments. The majority of isolates (97.6%) belonged to clonal complexes CC2 and CC87 of E. faecalis. All isolates were resistant to vancomycin and teicoplanin, and resistance to ciprofloxacin and aminoglycosides (high level) was very prevalent in this group. VanA phenotype was associated with 16 types of Tn1546, carrying insertion sequences IS1216, ISEfa4, IS1251 and IS1542, located on repUS1pVEF1, rep1pIP501, rep2pRE25, rep9pAD1/pTEF2/pCF10 and rep6pS86 replicons. The most common Tn1546 B- and BB-type transposons, harbouring one or two copies of IS1216, were inserted between rep18ap200B and repUS1pVEF1 genes and located on ~ 20 kb and 150–200 kb plasmids. VREfs-VanA in Poland represent a polyclonal group, indicating a number of acquisitions of the vanA determinant. The repUS1pVEF1-vanA plasmids, unique for Poland, were the main factor beyond the acquisition of vancomycin resistance by E. faecalis, circulating in Polish hospitals.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Enterococcus faecalis is the most common species of enterococci, widely distributed in humans, animals and the environment. It also significantly contributes to the overall number of healthcare-associated infections (HAIs) caused by antimicrobial resistant bacteria [1]. Population analyses of hospital E. faecalis based on multilocus sequence typing (MLST) revealed the existence of a few clonal complexes (CCs) dominating among clinical isolates [2,3,4,5]. Three of these complexes, CC2, CC9 and CC87, include invasive, multi-drug resistant isolates frequently obtained from hospitalized patients but found also in faecal carriage [6].
The majority of antibiotics, including aminoglycosides, cephalosporins, fluoroquinolones, tetracyclines, phenicols, macrolides, lincosamides and streptogramins, are often excluded from therapy of E. faecalis infections due to various intrinsic and acquired resistance mechanisms [7]. Thus, glycopeptides (vancomycin and teicoplanin) represent important drugs in the treatment of enterococcal infections. Resistance to these compounds in E. faecalis is being reported in Europe, although with much lower frequency (1.1%) than in Enterococcus faecium (19.0%) [8]. Recently, in Poland, 2.5% of all E. faecalis invasive infections are caused by vancomycin-resistant E. faecalis (VREfs) [1]. The appearance of VREfs poses a therapeutic challenge and a risk for transmission of glycopeptide-resistance to other hospital pathogens, in particular to Staphylococcus aureus [9, 10].
The vanA gene cluster, which confers resistance to both vancomycin and teicoplanin (VanA phenotype), is predominantly associated with different variants of Tn1546-type transposons, which result from point mutations, deletions and activity of insertion sequences [11,12,13]. In Poland, the first VREfs isolate with the vanA determinant was observed already in 1998 [14]. Two plasmid families associated with VanA determinants have been described for E. faecalis, including broad host Inc18 plasmids and pheromone-responsive plasmids, the latter restricted almost exclusively to this species [10, 15, 16]. Multiple acquisitions and occasional losses of vanA-carrying mobile genetic elements (MGEs), variation of Tn1546 insertion sites and clonal expansion of particular strains often following vanA acquisition describe the dynamics of hospital VREfs [17]. However, the knowledge about worldwide diversity and activity of VanA-MGEs in E. faecalis and the global population structure of VREfs is still far from being complete.
After the first report of VREfs-VanA in hospital settings in Poland in 1998 [14], these pathogens re-appeared in 2004 and since then are regularly submitted to the National Reference Centre for Susceptibility Testing (NRCST). The aim of this study was to provide the in-depth phenotypic and molecular characteristics of VREfs-VanA clinical isolates from 2004 to 2016 collected by the NRCST, with the special focus on its population structure and vanA-associated MGEs in order to better understand the epidemiology of these pathogens in our country.
Materials and methods
Bacterial isolates and phenotypic testing
The study comprised 125 consecutive, non-repetitive (1 isolate per patient) VREfs-VanA isolates received by the NRCST from 38 hospitals in 25 cities in Poland over the period 2004–2016, including fifteen isolates from year 2004 [3, 18] and the 1207/14 isolate from 2014 [19]. Twelve isolates (9.6%) were from invasive infections (all isolates from blood), 61 isolates (48.8%) were from non-invasive infections (23, 15 and 23 isolates from urine, wounds and other sources, respectively) and 52 isolates (41.6%) represented faecal carriage. Additionally, the first Polish ST87 VREfs-VanA isolate from 1998 [14] was used for comparative purposes. Presumable outbreaks were defined as appearance of two or more VREfs-VanA infections in a single hospital at the same or narrow time [20]. Antimicrobial susceptibility was tested using gradient tests for daptomycin, teicoplanin and vancomycin (BioMérieux, Marcy-l’Etoile, France), and by a broth microdilution method for the remaining compounds (ISO 20776–1 standard). The results were interpreted following the European Committee on Antimicrobial Susceptibility Testing (EUCAST)-approved breakpoints for ampicillin, ciprofloxacin, gentamicin, streptomycin, vancomycin, teicoplanin, tigecycline and linezolid [21], and the Epidemiological Cut-Off (ECOFF) values for compounds without defined breakpoints, such as penicillin, tetracycline, daptomycin and chloramphenicol (http://mic.eucast.org/Eucast2/, last accessed 10th March 2022). The isolates were examined for the production of cytolysin using a hemolytic test [22]. In brief, bacterial cultures were streaked on THB agar with 5% of horse blood, incubated 24–72 h in 5% CO2 at 37 °C and examined for the presence of clearing zones around the colonies. The production of aggregation substance (AS) was assessed by observation of cell clumping in the presence of sex pheromone from culture supernatants of the OG1X strain of E. faecalis [23].
DNA isolation and PCR detection of van determinants
Total DNA of isolates was extracted using the Genomic DNA Prep Plus kit (A&A Biotechnology, Gdansk, Poland). The detection of the vanA and vanB genes was performed by PCR [24, 25] with E. faecium BM4147 and E. faecalis V583 as positive controls, respectively.
Genotyping of isolates by MLST and PFGE
MLST was performed as described [26] using the web-accessible database https://pubmlst.org/organisms/enterococcus-faecalis (last accessed 18th October 2021) to establish alleles and sequence types (STs) [27]. STs were grouped into CCs by the comparative eBURST analysis performed against the whole E. faecalis MLST database (https://pubmlst.org; last accessed 31st March 2020). The MLST data for three isolates (6210/09, 6432/09 and 6878/09) were reported previously [28] and used in the current analyses. Pulsed-field gel electrophoresis (PFGE) was performed according to de Lancastre et al. [29] for agarose plugs preparation, followed by the procedure of Clark et al. [30] for total genomic DNA purification. Purified DNA in plugs was digested with the SmaI restriction enzyme (Fermentas, Vilnius, Lithuania). Electrophoresis was performed at 14 °C for 22 h with pulse time 1–30 s at 6 V/cm2 in 0.5 × TBE buffer. Lambda PFG Ladder (New England Biolabs, Ipswich, MA) was used as a DNA size standard. The Dice similarity coefficient (1% optimization, 1% tolerance, 1% tolerance change) and the unweighted pair-group mean arithmetic method (UPGMA) in the Bionumeric software (Applied Maths, Kortrijk, Belgium) were used to analyse PFGE-banding patterns, with the 85% similarity cut-off value defining a PFGE type (PT).
Tn1546 typing and localization
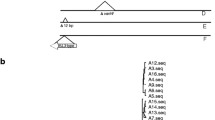
Tn1546 transposon structure was investigated by PCR mapping and sequencing (Supplementary Table 1 and references therein) of selected regions encompassing 7571 bp out of 10,851 bp, i.e. ~ 70% of the transposon (Fig. 1.). The Tn1546 sequence of E. faecium BM4147 (GenBank acc. no.: M97297) [11] was used as a wild-type reference (A1 type). The nomenclature of Tn1546-type transposons in the current study was based on the alphanumeric code used previously for Polish E. faecium VanA [31], according to which the ‘A’ types referred to transposon variants affected only by point mutations; the ‘B’ types contained 1–2 copies of IS1216 (B and BB types); the C, E and J types carried IS1251, ISEfa4 and IS1542 elements, respectively. Transposons with more than one IS type were described by a two- or three-letter code (e.g. ‘BC’ for transposon with both IS1216 and IS1251). The Arabic numerals indicated the presence of point mutations compared to the wild-type A1 transposon and/or differences in orientation of ISs and the localization of their insertion sites (e.g. B2-B9). Primers used for PCR targeting junctions between Tn1546 and its insertion sites were designed based on 1207/14 isolate [19] and genomic sequences obtained in this study (Supplementary Table 1).


Diversity of Tn1546 transposon types among E. faecalis VanA isolates. Black arrows, transposon genes; stars, positions of point mutations; areas of the transposon analysed by PCR mapping and sequencing shadowed; the A5912G and A10086G point mutations detected on Illumina reads; dashed lines, deletions in the left arm of the transposon; grey arrows, ISs; DR, direct repeats; ins, insertion; del, deletion; n.d.,– not determined; a number of isolates, if larger than one, are given in brackets; b reference Tn1546 (M97297). Nucleotide positions correspond to the reference Tn1546 transposon
Plasmid analysis
Detection of 19 rep families and the unique reppMG1 gene was performed according to Jensen et al. [32]. PCR primers for amplification of repUS1pVEF1 and rep18ap200B not included in the original scheme were designed based on the 1207/14 genome [19]. PCR detection of asa, cylA, bee, reppLG1, repUS1pVEF1, rep18ap200B, plasmid addiction systems and relaxase genes was performed as described (Supplementary Table 1 and references therein). A few randomly selected PCR products were sequenced to confirm the results for each of the detected genes. For plasmid profiling and hybridization analyses, DNA in agarose plugs was obtained as described above [28, 29], treated with S1 nuclease (Takara Bio, Japan) and separated by PFGE [33] followed by blotting onto the Hybond membrane (GE Healthcare, Buckinghamshire, UK) by a capillary transfer. The size of plasmid bands was estimated according to Lambda PFG Ladder and plasmids < 50 kb were additionally compared to the 21.6 kb p1207_4 plasmid of the 1207/14 isolate [19]. Hybridization was carried out using the Amersham ECL Random-Prime Labelling and Detection System (GE Healthcare) with the vanA, rep1pIP501, rep2pRE25, rep6pS86, rep8pAM373, rep9pAD1/pTEF2/pCF10, rep13pC194, rep18bpEF418, repUS1pVEF1, par, relpAD1, relpCIZ2, relpAMalpha, asa and cylA probes.
Conjugation
Mating experiments were performed with E. faecalis OG1RF and E. faecium 64/3 recipients according to the procedure developed for strains with a low transfer efficiency [34]. To this end, overnight liquid cultures of donors and recipients were mixed in a 1:1 ratio, spread on BHI agar plates and incubated at 37 °C for 24 h. Bacterial cells were then transferred onto a selective medium (BHI agar with 32 mg/l vancomycin, 64 mg/l fusidic acid and 64 mg/l rifampicin) and incubated at 37 °C for 48 h.
Whole-genome sequencing and data analysis
For whole genome sequencing (WGS), total DNA of selected isolates was obtained using the Genomic Mini AX Bacteria Kit (A&A Biotechnology, Gdynia, Poland) according to the manufacturer’s instructions and sequencing was carried out on the Illumina MiSeq Platform with the PE300 mode (Illumina Inc., San Diego, CA) as an external service (Genomed S.A., Warsaw, Poland). Reads were trimmed with Cutadapt v 1.16 [35], assembled using Spades v 3.11.1 [36] and annotated with PROKKA 1.11 [37]. Supplementary manual BLASTx analyses (https://blast.ncbi.nlm.nih.gov/) were used when appropriate. Complete genomic sequences of E. faecalis were downloaded from GenBank (23rd February 2022), analysed using mlst [https://github.com/tseemann/mlst; 8th March 2022 date last accessed] and annotated with PROKKA. Core genome alignments were obtained with Roary [38] and used in RAxML [39] for construction of Maximum Likelihood (ML) trees. For identification of vanA-plasmids of E. faecalis in GenBank the complete Tn1546 sequence was used as a query for blastn search of the nt/nr database (as of 29th October 2021), limited by: organism: “Enterococcus faecalis (taxid:1351)” and the hits with the query coverage over 50% were included in the final set. The PlasmidFinder 2.0.1 [40] and ResFinder 3.0 [41] services (both last accessed 25th February 2022) were used to identify known plasmid replicon families and resistance genes, respectively, in genomic data. Conjugation transfer-associated regions were detected by oriTFinder [42]. The p1207_4 plasmid sequence was visualized using the BLAST Ring Image Generator (BRIG, http://brig.sourceforge.net) [43].
Statistical analysis
Chi-squared test was used to assess the differences of distributions, with p ≤ 0.05 considered significant.
Accession numbers
The whole-genome shotgun project has been deposited at DDBJ/ENA/GenBank under the BioProject number PRJNA731638. The accession numbers of draft genome sequences described in this paper are listed in Table 2. The complete genome sequence of 1207/14 isolate of VREfs-VanA published recently [19] can be found in the DDBJ/ENA/GenBank under the accession numbers CP075604 (chromosome), CP075605 (p1207_1), CP075606 (p1207_2), CP075607 (p1207_3), CP075608 (p1207_4), CP075609 (p1207_5) and CP075610 (p1207_6).
Results
Population structure of Polish VREfs-VanA
During the study period (2004–2016), two cities, Warsaw and Poznań, were most affected by VREfs-VanA, with 86 isolates (68.8%) originating from 14 different medical centres and causing several outbreaks (outbreaks A, C, D1-D3, E, G, H and I in Fig. 2). The remaining 39 isolates were responsible for four small outbreaks in Pl, Je, Bp and Si hospitals (outbreaks B, F, J and K, respectively) as well as 26 sporadic cases in other Polish cities. MLST and PFGE discerned nine STs and 23 different PTs within the studied population, respectively (Fig. 2.). In the MLST analysis, the vast majority of isolates belonged to two clonal complexes, CC87 (n = 71; mainly STs 87 and 28) and CC2 (n = 51; solely ST6). Three isolates belonged to distinct STs 16 and 215. In the PFGE analysis, six most prevalent PTs such as 1, 2, 16, 19, 20 and 21 were characteristic for 100 isolates (80.0%) and grouped 70 out of 76 (92.1%) isolates presumably involved in outbreaks. The most prevalent PT1 (n = 24) was associated with ST87 (n = 17) and ST464 (n = 7) isolates, both belonging to CC87. Other numerous PTs also highly correlated with specific STs, i.e. PT2 corresponded solely to ST87, PT16 and PT19 were present among ST6 isolates while PT20 and PT21 belonged exclusively to ST28.


PT-based dendrogram of VREfs-VanA in Poland (n = 125) collected during 2004–2016. Bp, Biała Podlaska; El, Elbląg; Gd Gdańsk,; Gr, Grajewo; Gro, Grodzisk Mazowiecki; Gry, Gryfice; Je, Jelenia Góra; Ka, Katowice; Lo, Łódź; Lu, Lublin; My, Myślenice; Op, Opole; Ost, Ostrów Mazowiecki; Ot, Otwock; Pl, Płock; Ple, Pleszew; Po, Poznań; Rz, Rzeszów; Si, Siedlce; So, Sosnowiec; Sw, Świdnica; Sz, Szczecin; To, Toruń; Wa, Warszawa; Wr, Wrocław; Zi, Zielona Góra; the city abbreviation is followed by the centre number; presumable outbreaks designated with letters A-K; TgR, isolate resistant to tigecycline; ChR, isolate resistant to chloramphenicol; CpS, isolate susceptible to ciprofloxacin; not HLSR, isolate susceptible to high concentration of streptomycin; not HLGR, isolate susceptible to high concentration of gentamicin; only phenotypes that differentiate the isolates are shown. Filled and empty circles indicate strong and weak haemolysis activity, respectively. The first Polish VREFs-VanA isolate (7946/1998) included for comparative purposes
The first Polish VREfs-VanA was isolated in a Gd medical centre in 1998 [14] and belonged to CC87 (ST87/PT6). VREfs-VanA remained then absent in Poland until 2004, when the first CC87 outbreak occurred in the Wa-10 medical centre, involving five ST87/PT1 isolates (outbreak A in Fig. 2). Single ST87/PT1 isolates were obtained in the same year also from two other centres (Ost, Gro). Another outbreak of ST87/PT1 VREfs-VanA (7 isolates) took place in the Po-2 medical centre also in 2004 (outbreak C). The ST87/PT2 isolates (n = 17) caused outbreaks in 2010–2011 in the Wa-2 and Wa-8 medical centres (outbreaks D1 and D2) and occasionally ST87/PT2 VREfs-VanA appeared also in Wa-9, Wa-13 and Gro hospitals until 2014. In parallel, a multicenter spread of ST28 isolates (n = 18 isolates) belonging to related PT20 and PT21 caused outbreaks in the Wa-1, Bp and Si hospitals during 2007–2009 (outbreaks I, J and K).
While CC87 predominated in Polish hospitals during 2004–2010 (52 out of 56 isolates from this period), isolates belonging to CC2/ST6 constituted the majority of VREfs-VanA isolates during the next six years (46 out of 68 isolates from 2011 to 2016). The first, sporadic ST6/PT19 isolate was observed in the Wa-8 medical centre in 2011 and likely this strain was transferred to Wa-2, where representatives of ST6/PT19 were responsible for the large hospital outbreak H (19 isolates) in the 2011–2012 period. CC2 isolates were also detected in other centres in Warsaw as well as in other cities during 2007–2016, in association with PT15 (n = 3; the outbreak F in Je), PT16 (n = 16) and several sporadic PTs (Fig. 2). The group of ST6/PT16 isolates was associated with sporadic isolations except for a small outbreak G (2 isolates) in the Po-7 medical centre in 2013.
Phenotypic characteristics of the collection and main CCs
The MIC values of glycopeptides ranged from 128 to > 256 mg/l for vancomycin and from 8 to > 256 mg/l for teicoplanin in the studied population. All isolates carried vanA and lacked vanB. Resistance to ciprofloxacin, high level gentamicin and high level streptomycin resistance were very common in the whole collection (99.2%, 96.0% and 97.6%, respectively). Three isolates were resistant to tigecycline. All the isolates were susceptible to ampicillin and linezolid, and exhibited MIC values below the ECOFFs for penicillin and daptomycin. The MIC values for tetracycline and chloramphenicol were above the corresponding ECOFFs in the case of all isolates and 46 (36.8%) isolates, respectively (http://mic.eucast.org/Eucast2/, last accessed 15th July 2020). Haemolysis was typical for 63 isolates (50.4%), among which 48 isolates displayed a strong haemolysis and 15 isolates displayed a weak haemolytic activity (Fig. 2). All but two haemolytic isolates carried the cylA gene (Fig. 2). The AS gene (asa) was detected in 108 isolates (86.4%); however, only eight isolates were positive in clumping test in the culture supernatant of the OG1X strain of E. faecalis. Two main CCs, 6 and 87, did not show significant differences concerning antimicrobial susceptibility phenotypes except chloramphenicol resistance, which was more prevalent in CC87 (59.7% of this CC, compared to 5.9% of CC2, p < 0.0001). The majority of isolates with strong haemolysis belonged to CC87 (n = 44; p < 0.0001), while weak haemolysis was associated with CC2 (n = 14; p = 0.0007). Haemolysis-negative isolates (n = 62) belonged to both clonal complexes and the presence of cylA was observed for 22 of these isolates, including mainly the outbreak H (Fig. 2).
Tn1546-type transposon diversity
Tn1546-typing distinguished seven transposon types A, B, BB, BC, BJ, BBJ and E, which included 15 subtypes (Fig. 1). The vast majority, i.e. 114 isolates carried Tn1546 of various B-types, disrupted by (i) one or two copies of IS1216 (subtypes B2, B4-B9 and BB4-BB6); (ii) IS1216 and IS1251 (subtype BC6); and (iii) IS1216 and IS1542 (subtypes BJ and BBJ). Eight isolates carried E-type with ISEfa4 insertion and a single isolate possessed transposon without ISs, differing from the wild-type Tn1546 by the presence of a point mutation (the A7 subtype). Apart from the presence of various ISs, the variability of Tn1546 was also associated with the presence of point mutations, insertions and deletions. The IS1216 activity caused deletions of different size in the regions adjacent to the IS insertion site in the majority of transposon types (B2-B9, BB4-BB6, BC6, BJ, BBJ). Five different nucleotide substitutions (G7747T, T7658C, G8234T, G9063T, C9692T) described previously [31, 44,45,46] and two novel ones (A5912G, A10086G) were present in subtypes B6, B9, BC6 and E. Insertion/deletion of a single T nucleotide within the poly-T tract in the vanY gene (nt 9063–9071) occurred in the B4, B5, B6, B7 and B9 subtypes, resulting in translational frameshifts and a truncated VanY. For a single isolate, Tn1546 type could not be defined due to problems with PCR amplification of parts downstream of the vanA gene.
The first VREfs-VanA from 1998 harboured the BB3-type Tn1546, not observed in any later isolate. BB4- and E-types of Tn1546 were typical for isolates causing the early CC87 outbreaks A and C (Fig. 2) in the Wa-10 and Po-2 medical centres, respectively. The most prevalent subtype B5 (84 isolates, 67.2%) was present in isolates belonging to both CC2 and CC87, occurring predominantly in Warsaw hospitals. B5 was associated with several outbreaks (B, D1-D3, E, H, I, J, K in Fig. 2) as well as with sporadic isolations. The second most prevalent subtype B4 (13 isolates) varied from B5 by a single T insertion within the poly-T tract in the vanY gene and was typical for sporadic cases in different cities during the 2008–2013 year period. The remaining Tn1546 types (1–2 isolates/type) were sporadically detected among 14 isolates not involved in outbreaks.
VREfs-VanA plasmidome, vanA-plasmids, their transfer and epidemiology
PCR screening for plasmid replicon types was performed according to the original typing scheme for Gram-positive bacteria [32] and additionally included detection of repUS1pVEF1, repUS11pTEF3 and rep18ap200B. The most frequent plasmid replicons were rep9pAD1/pTEF2/pCF10, characteristic for pheromone-responsive plasmids, rep18ap200B of Rep_3 theta plasmids and repUS1 pVEF1 from the Inc18 group of plasmids (Table 1). The majority of rep genes was evenly distributed in both CC87 and CC2, with the exception of rep13pC194, exclusively associated with CC87, and rep6pS86, significantly overrepresented in this CC (38.0% of CC87 compared to 5.9% of CC2, p = 0.0001). The rep17pRUM gene was detected only in the first Polish VREfs-VanA isolate from 1998. Analysis of the distribution of plasmid mobilization and addiction systems showed the abundance of the MOBC2 relaxase gene (122 isolates) and the par addiction system (56 isolates), characteristic for the pAD1 pheromone-responsive plasmid [47, 48]. Other detected relaxase genes included two MOBP7 genes of pCF10 and pCIZ2 [48, 49], and the MOBV gene, typical for pAMalpha [48, 50] (9, 106 and 47 isolates, respectively).
Fifty isolates belonging to both main CCs 2 and 87 (22 and 26 isolates, respectively), as well as representing STs 16 and 215 (single isolates each) obtained from 32 medical centres and representing 21 PFGE types and 14 Tn1546 types/subtypes, were used in PFGE-S1 Southern-blot hybridization analyses and conjugative transfer experiments (Fig. 3). These isolates constituted 40% of the studied collection and were selected to maximally represent its diversity. The first Polish VREfs-VanA was additionally included in this group. Single vanA-plasmids were detected in 13 isolates, while 37 isolates carried two co-resident vanA-plasmids, resulting in a total number of 87 vanA-plasmids (Fig. 3A).


PFGE-S1 Southern blotting results for selected 50 VREfs-VanA and the first Polish VREFs-VanA isolate (7946/1998) (A) and comparative PFGE-S1 Southern blotting with the vanA probe for conjugation-positive isolates (donors D1-D15) and their E. faecalis and/or E. faecium transconjugants (B). Eight isolates analysed by WGS underlined; a city abbreviations and outbreak designation as in Fig. 2; isolates positive for conjugation in bold
The most prevalent repUS1pVEF1-vanA plasmids (n = 63) were detected in 37 isolates, of which 26 isolates had two repUS1pVEF1-vanA plasmid bands, ≤ 30 kb and 150–200 kb in size (Fig. 3A). The relpCIZ2 relaxase gene (MOBP7) was associated with the majority of repUS1pVEF1-vanA replicons (50 out of 63 plasmids). The repUS1pVEF1-vanA replicons were observed for outbreak isolates in Warsaw (A, I, D1, D3 and H) as well as in 12 other cities during the whole study period. Isolates with these plasmids carried mainly BB- and B-subtypes of Tn1546 transposon. rep2pRE25 was present altogether on 12 vanA-replicons, ranging from 20 to 160 kb in size. In six cases, rep2pRE25 was located on 30–50 kb repUS1pVEF1-vanA plasmids, described above. These putative rep2pRE25-repUS1pVEF1-vanA multireplicons appeared in 2011 and were associated with two small outbreaks in Wa-8 (D2) and Pl (B) and later they were also detected in Wa-7 and Po-6 sporadic cases in 2016. The rep1pIP501 gene was associated with four 80–270 kb vanA-plasmids, including one 100 kb rep2pRE25-vanA plasmid. The rep1pIP501-vanA plasmids were present among early isolates, mainly from the C outbreak in Po-2 in association with the E-type Tn1546 transposon.
Other vanA replicons were detected sporadically. In a single isolate rep2pRE25-repUS1pVEF1-vanA plasmid hybridized also with rep9pAD1/pTEF2/pCF10 gene. Apart from this isolate, rep9pAD1/pTEF2/pCF10 was also specific for two large vanA-plasmids, 300 kb and 360 kb in size. The 360 kb rep9pAD1/pTEF2/pCF10-vanA plasmid carried additionally rep6pS86. A single isolate carried 80 kb vanA-plasmid with both rep6pS86 and rep2pRE25 replication genes. The first Polish VREfs-VanA isolate and the remaining 11 isolates from the collection harboured vanA-plasmids ranging from 20 to 270 kb in size; however, their replicon type could not be determined (Fig. 3A).
Fifteen isolates out of 50 selected for Southern-blotting were positive for conjugative transfer of vanA determinants to a susceptible recipient (Fig. 3B). Six of them transferred vanA plasmids to both E. faecalis OG1RF and E. faecium 64/3 recipient, seven only to E. faecium and two only to E. faecalis. The most commonly transferred vanA plasmids belonged to repUS1pVEF1 replicons of ≤ 30 kb and 150–200 kb in size (9 isolates out of 36 isolates with repUS1pVEF1-vanA plasmids investigated, Fig. 3A) and for eight of them, no significant changes in plasmid sizes were observed during conjugation (Fig. 3B). Additionally, two rep2pRE25-repUS1pVEF1-vanA plasmids (30–50 kb), a single rep2pRE25-rep9pAD1/pTEF2/pCF10-repUS1pVEF1-vanA plasmid (50 kb) and a single rep6pS86-rep9pAD1/pTEF2/pCF10-vanA plasmid (360 kb) were transferable by conjugation, however with significant changes in plasmid size (Fig. 3B). Transfer was also achieved in the case of two isolates harbouring vanA-plasmids with unknown replicon types.
To compare the distribution of rep genes associated with vanA-plasmids in our collection, a set of 22 vanA-plasmids from E. faecalis reported from other countries was assembled (Supplementary Table 2). The majority of these isolates was obtained from human sources in various countries during 1996–2016. The vanA-plasmids in E. faecalis varied in size from 31.4 kb to 128 kb and harboured rep1, rep2, rep7a, rep9b, rep9c, repUS1 and repUS43, with one to three rep genes per plasmid. Only three plasmids, 31.4, 76.0 and 107.6 kb in size, harboured repUS1, characteristic for the majority of vanA-plasmids from the current study. The co-localization of rep18a with repUS1pVEF1 on vanA-plasmids, common among Polish VREfs-VanA (see below), was not observed elsewhere. Six plasmids in the reference set represented multireplicons with various combinations of rep genes. The rep9pAD1/pTEF2/pCF10-vanA plasmids, which were the most common ones in the reference set, were observed only sporadically in our collection (12 vs. 3 plasmids, respectively).
WGS analysis of selected VREfs-VanA
The complete genome of 1207/14 isolate was reported previously [19] and the Illumina MiSeq sequencing of seven VREfs-VanA isolates and three transconjugants was performed in the current study (Table 2). Selection of isolates for WGS analysis was aimed at providing the best representation of STs, Tn1546 types and vanA-plasmid PFGE-S1 profiles detected in the studied population, including isolates with the most common characteristics as well as those appearing sporadically (Figs. 2 and 3). In particular, WGS was carried out for three isolates harbouring the most prevalent B5-type Tn1546 in various backgrounds (STs 6, 28, 87), an isolate with the related B9-type in ST16, not belonging to any of the two predominant CCs, an isolate with BC6-type in ST464 (CC87) and single isolates with the A7 and E transposon types associated with the ST6 and ST87, respectively. Additionally, the first Polish VREfs-VanA isolate was also included in WGS.
The 1207/14 isolate demonstrated the most common ≤ 30 kb and 170 kb repUS1pVEF1-vanA plasmid profile and represented one out of four isolates transferring vanA to both E. faecium and E. faecalis without any noticeable change in size and number of vanA-plasmids (donor D10, Fig. 3B). This isolate contained five plasmids (p1207_1-p1207_5, including the 21.6-kb plasmid p1207_4 harbouring the vanA operon) with 10 known rep genes, and a small 2-kb plasmid with an unknown rep type [19]. The analysis in the current study revealed that p1207_4 carries 24 probable protein-coding genes (Fig. 4), including three rep genes: (i) repUS1pVEF1 (Inc18) with 100% identity to the rep gene of pVEF1, pVEF2 and pVEF4 plasmids of E. faecium [51], (ii) rep18ap200B (Rep_3) with 99.98% identity to the repA gene of p200B of E. faecium [52] and (iii) a truncated repB with 100% identity to the repB gene present in several plasmids of E. faecium, e.g. in the ISMMS and E39 isolates [53, 54]. Screening the p1207_4 sequence for conjugation transfer-associated regions by oriTFinder revealed the presence of a mobilization gene belonging to MOBP7 family (relpCIZ2); however, no oriT was detected. The p1207_4 represented a unique mosaic plasmid structure, composed of two segments separated by IS1216. The 8.9 kb segment containing partial repB, rep18ap200B, rel, mobC, the Fst toxin gene and a set of genes, presumably involved in the biosynthesis of a putative bacteriocin belonging to the lactococcin 972 family, demonstrated 99.5% identity to the ISMMS_VRE_p3 plasmid of E. faecium [53]. The structure of the other, a 10.7 kb segment, which included a partial Tn1546 and the prgN, repUS1pVEF1 and parA genes was unique in the GenBank.

Map of the p1207_4 vanA-plasmid (GenBank accession no. CP075608); positions and directions of predicted coding sequences are indicated by arrows, with antimicrobial resistance genes in red, plasmid replication genes in blue, genes associated with conjugative transfer, mobilization and transposition in green and other CDSs in black
Illumina MiSeq sequencing of seven VREfs-VanA isolates demonstrated that in four isolates (3124/08, 5274/12, 753/12 and 5208/13) both vanA and repUS1pVEF1 reside on identical contigs, approximately 11 kb in size. As described above, these isolates carried repUS1-vanA plasmids of approximately 20 and 170 kb (Fig. 3A), and assembling sequencing reads of these four plasmids to isolate 1207/14 plasmids as reference confirmed the presence of p1207_4 sequences in all four isolates (Table 2). In the case of 5208/13, mapping Illumina MiSeq reads revealed the presence of the complete sequences of p1207_2 and p1207_3 in addition to p1207_4 (Table 2). The p1207_2 plasmid (70.1 kb) is an Inc18-plasmid, harbouring repUS11pTEF3 and conjugative transfer functions, while p1207_3 is a 46.5-kb plasmid with rep9apAD1 [19]. A single isolate (3300/04) exhibited the presence of approximately 20 kb vanA/rep1pIP501 contig, consistent with the previous Southern blotting results (Fig. 3A), and mapping Illumina reads of 3300/04 revealed the presence of complete sequences of p1207_3 (Table 2). The vanA-contigs of three remaining isolates (7946/98, 574/14 and 1739/15) contained no plasmid replication genes (Table 2). Mapping Illumina reads of 574/14 and 1739/15 isolates revealed the presence of p1207_2 and p1207_1 plasmid sequences in these isolates, respectively.
Three transconjugants, 1207/14TKEfs, 5208/13TKEfm and 574/14TKEfm, were selected for Illumina MiSeq sequencing. Mapping sequencing reads of 1207/14TKEfs to the plasmid sequences from the 1207/14 donor confirmed the presence of the p1207_4 plasmid in 1207/14TKEfs and additionally revealed acquisition of the p1207_1 plasmid by this transconjugant. p1207_1 is the largest plasmid found in 1207/14 (75.1 kb) and carries four rep genes (rep9bpEF62pC, rep6pS86, rep7apRE25 and repUS12SAP014A), conjugation transfer genes and antimicrobial resistance determinants aph(3')-III, ant(6)-Ia, erm(B) and cat [19]. In the case of 5208/13 transconjugants, the change of the original repUS1pVEF1-vanA plasmid profile (20 kb and 170 kb) was observed (a single 170 kb repUS1pVEF1-vanA plasmid in 5208/13TKEfm and a single 110 kb repUS1pVEF1-vanA plasmid in 5208/13TKEfs, respectively, as shown in Fig. 3B). Mapping Illumina MiSeq reads revealed the presence of the complete sequences of p1207_2 in addition to p1207_4 in 5208/13TKEfm transconjugant (Table 2). For the 574/14 isolate transfer of vanA to E. faecalis OG1RF was not achieved, while the 574/14TKEfm transconjugant carried a single 120 kb vanA-plasmid, identical to the vanA-plasmid band observed for the donor (Fig. 3B). Mapping Illumina reads revealed the presence of complete p1207_2 sequence in 574/14TKEfm (Table 2). Plasmid replication genes typical for p1207_1, p1207_2 and p1207_4 were the only rep genes present in the analysed transconjugants, according to the PlasmidFinder results.
Relationships of CC87 and ST6 isolates with isolates from other countries
Genomic sequences of six Polish isolates belonging to CC87 and two Polish isolates of ST6 were used for joint analyses with genomic sequences available at GenBank. The reference set for CC87 consisted of 23 isolates, originating from 12 other countries and obtained during 1986–2020 from various sources (Supplementary Fig. 1A). The Roary analysis identified 6203 genes, of which 2295 represented the core genome. In the ML tree (Supplementary Fig. 1A), three Polish ST87 isolates, including the first Polish VREfs-VanA from 1998 and two isolates from 2004 and 2013, formed a group separated from other Polish CC87 isolates. Instead, these three isolates clustered with the majority of ST28 isolates from other countries. Although the three isolates appeared very closely related in the core-genome analysis, each of them had a unique content of plasmid rep genes and resistance determinants, and carried the Tn1546-type transposon in a different genetic context (see below), suggesting an independent acquisition of the vanA determinant. Two Polish isolates of ST464 and a single Polish ST28 isolate formed two separate branches, unique for Poland. Two ST464 isolates were very closely related in the core-genome analysis and shared antimicrobial resistance determinants but demonstrated a different genetic context of Tn1546 (see below). For the analysis of ST6 isolates, 155 genomic sequences were retrieved from GenBank. To improve the clarity of presentation, very closely related or indistinguishable isolates were removed from an initial ML tree (data not shown) and such clusters were represented by a single randomly chosen isolate (Supplementary Fig. 1B). The final ML tree included 33 reference sequences, characteristic for human isolates obtained during 1986–2019 from 10 countries. The pangenome in this group consisted of 6375 genes, of which 2048 belonged to the core genome. Two Polish isolates were closely related in this analysis and belonged to a larger cluster, grouping several vancomycin-resistant isolates from the USA from the second half of the 2010s as well as single isolates from the UK and Denmark. Both CC87 and ST6 isolates demonstrated the abundance of resistance determinants (up to 8 and 7 genes per isolate in CC87 and ST6 groups, respectively) and plasmid rep genes (up to 8 genes per isolate in both groups). The most ubiquitous genes included tet(M), erm(B) and aminoglycoside high-level resistance genes as well as rep9a and rep9b, characteristic for pheromone-responsive plasmids. The rep18ap200B and repUS1pVEF1 genes flanking Tn1546 in the majority of isolates in our collection (see below) were observed solely for three Polish isolates from CC87 and a single Polish isolate of ST6. No particular associations between the distribution of resistance genes or rep genes with the ML tree groupings were found.
The genetic context of Tn1546
The analysis of Tn1546 flanking regions on p1207_4 plasmid [19] revealed that rep18ap200B and partial repB genes were present upstream and the prgN and repUS1pVEF1 genes were located downstream of the transposon. This type of Tn1546 flanks was designated rep18a-repUS1 and was not as yet observed elsewhere (GenBank nr/nt database, 23rd June 2022, date last accessed). The same genetic neighbourhood downstream Tn1546 was found also in vanA/repUS1pVEF1 contigs of four isolates analysed by WGS (see above). However, regions upstream Tn1546 were lacking from these contigs, consistent with the presence of IS1216 in B5- and B9-type transposons in these isolates, which precluded assembly of this region from Illumina MiSeq reads. The investigation of Tn1546 insertion sites in the remaining four isolates analysed with the Illumina MiSeq sequencing allowed to define the following Tn1546 flanks: (i) R-rep1 with the rep1pIP501, res and topoI genes downstream of Tn1546; (ii) relE-merA with the relE toxin gene upstream of Tn1546 and the merA gene downstream of Tn1546 and (iii) R-arsR with the arsR gene downstream of transposon (Fig. 1, Table 2). The R-rep1 flank was not reported previously. The flank designated relE-merA was described recently in three vanA plasmids (pVB096, pVB039 and pVBR48) from E. faecium ST133 isolates [55]. The R-arsR flanking sequence, typical for the first Polish VREfs-VanA isolate, was commonly detected in E. faecium genomes (116 hits in GenBank) while only a single E. faecalis plasmid pR712_01 with an identical flank was deposited in GenBank (CP036247.1).
Based on the sequences of four types of flanks described above, PCR primers specific to junctions between plasmids and Tn1546 were designed (Supplementary Table 1) and used for screening the whole collection (Figs. 1 and 2). A hundred-nine isolates (87.2%) had the repUS1 downstream flank but only 92 of them yielded the PCR product of the expected size for the rep18a upstream flank. Four of the remaining isolates yielded smaller PCR products due to either deletion within partial repB (3 isolates, the flank designated ΔrepB-repUS1) or deletion encompassing a fragment of rep18ap200B and almost entire repB gene (1 isolate, the flank designated Δ[rep18a-repB]-repUS1). For seven isolates obtained from various Warsaw medical centres during 2014–2016, the upstream flanking region was amplified with the use of a new primer rep18a_up_new, located ~ 500 bp upstream of the previously used primer rep18a_up, revealing the presence of ISEfa4 insertion within rep18ap200B (the flank designated ISEfa4-repUS1). For seven remaining isolates, the left flank could not be defined and these flanking sequences were designated R-repUS1. Among the remaining 17 isolates negative for the rep18a-repUS1 flanks, eight outbreak isolates from the Po-2 medical centre in 2004 (Tn1546 type E) showed the presence of R-rep1, four unrelated isolates carried R-arsR, while relE-merA remained typical only for the 574/2014 isolate, analysed previously by WGS. In the case of four remaining unrelated isolates, the Tn1546 insertion site could not be identified.
Distribution of different Tn1546 flanking regions in the studied population highly correlated with the results of Tn1546 typing (Figs. 1 and 2). Isolates with the rep18a-repUS1 flanks were prevalent during the entire study period and these flanks were associated mainly with the common B5, B4 and BB4 subtypes of Tn1546. ISEfa4-repUS1 flank, associated with B5 and B4 subtypes, was detected during the Wa-12/E outbreak and among sporadic isolates in two other Warsaw medical centres during 2014–2016. The R-rep1 flanking sequence was typical for E-type transposon in the C outbreak isolates from Po-2. The R-arsR flanking region, besides of being typical for the first VREfs-VanA isolate, appeared sporadically in 2015–2016 in association with BC6 and BBJ transposon types. Thirty-one of 39 isolates carrying repUS1pVEF1 downstream Tn1546 demonstrated the characteristic plasmid profile with ≤ 30 kb and 150–200 kb vanA plasmids in PFGE-S1/hybridization with the repUS1 probe (Fig. 3).
Discussion
The results obtained in this study allowed identifying two clonal complexes, CC2 and CC87, as the ones being mostly responsible for an increasing prevalence of VREfs-VanA in Poland. Early studies showed that both these CCs were characterized by multi-drug resistance and an increased pathogenicity potential [4, 5, 25]. These findings were further supported by genomic analyses of 168 isolates of E. faecalis from the UK, among which three major lineages L1 (corresponding to CC2), L2 (corresponding to CC87) and L3 demonstrated a strong enrichment in several virulence and antimicrobial resistance genes, including van genes [17]. CC2 is dispersed globally, while CC87 is observed in Europe (including Poland), Asia and Africa [3,4,5, 56; https://pubmlst.org/organisms/enterococcus-faecalis, 27th October 2021 date last accessed]. In the current analysis, we noticed a significant replacement of CC87 by CC2 after 2010 and a similar time-trend in the distribution of the L1 and L2 lineages of E. faecalis occurred in British hospitals, where CC87/L2 practically disappeared after 2006 [17]. Both CC2/L1 and CC87/L2 include vancomycin-susceptible as well as vancomycin-resistant isolates [3, 5, 17], consistent with multiple acquisitions of van determinants by strains of these CCs.
Tn1546 transposon is a basic genetic element responsible for vanA distribution in enterococcal populations and its genetic diversity serves as epidemiological marker to trace horizontal transfer of glycopeptides resistance among strains and dissemination of VRE-VanA within and among medical centres [11, 45, 57]. However, in contrast to the situation in E. faecium, the diversity of Tn1546 in E. faecalis remains much less studied. Among Polish VREfs, IS1216 seems to play a major role in shaping the Tn1546 structure. This IS was responsible for the formation of B-type transposons most commonly detected during the entire study period and insertion of IS1216 was associated with deletions of the Tn1546 left end. Mapping genomic sequencing reads to Tn1546 as a reference revealed several examples of similar structures of Tn1546, i.e. devoid of the transposase and resolvase genes, among all three main lineages of E. faecalis in the UK [17]. In the set of vanA-plasmids assembled from the GenBank in the current study (Supplementary Table 2), the majority of the observed Tn1546 structures (13 out of 22) represented the wild-type Tn1546. Variants similar to the B-type transposons found in our study were also present in human isolates from Brazil and Portugal. Insertion of IS1216 between vanX and vanY, characteristic for the B8, BB3, BB4, BB6, BJ and BBJ types in our collection occurred also in Tn1546 located on plasmids of E. faecalis from human and chicken samples from Korea [58]. Apart from IS1216, three other ISs, ISEfa4, IS1251 and IS1542 were sporadically detected within Tn1546 in our collection. These ISs did not occur in Tn1546 of E. faecalis from other countries; however, some other ISs, such as IS3, IS256 and IS1657, were found. B4- and B5-type transposons commonly observed in our study were also present among E. faecium-VanA from Polish hospitals (31 and unpublished results), suggesting an extensive genetic exchange of the vanA mobilome between E. faecalis and E. faecium. Moreover, the E-type Tn1546 with ISEfa4 characterized in the current study in E. faecalis occurred concomitantly in E. faecium in the same hospital Po-2, in both cases associated with the rep1 replicon [31] additionally supporting the possibility of exchange of this element among enterococci in the hospital settings, with a broad host range Inc18 replicon as its vector. Such interspecies transfer was already suggested for the first VRE outbreak in Poland in 1998 [14].
Tn1546-type transposons are usually carried on conjugative plasmids from the Inc18 and RepA_N families, which promote their horizontal transfer in bacterial populations [10, 15, 16, 59,60,61]. In the current study, the majority of vanA-replicons belonged to the Inc18 family, being associated mainly with repUS1 and occasionally with rep1 and rep2 while pheromone-responsive RepA_N plasmids played a much lesser role as vanA carriers. In our PFGE-S1-hybridization studies, the repUS1 gene was typically associated with ≤ 30 kb and 150–200 kb vanA-plasmids in the majority of isolates. Such a profile was observed in several PFGE types of both epidemic CCs 2 and 87 as well as in other STs such as 16 and 215, consistent with independent acquisitions of the vanA-plasmid. In the set of 22 reference VREfs vanA-plasmids (Supplementary Table 2), the majority was below 100 kb in size. The repUS1 replicons were observed in two animal isolates and single human isolate from diverse localizations (The Netherlands, New Zealand, Brazil); one of these plasmids harboured also rep9c. The repUS1pVEF1 gene was first described in pVEF1 and pVEF2 of E. faecium from human and poultry in Norway [51]. Although p1207_4, similarly to pVEF1 and pVEF2 lacked a conjugation system, it demonstrated the presence of a relaxase gene and could be mobilized, presumably by conjugative plasmids present in the same bacterial cell, such as a pheromone responsive plasmid p1207_1, co-transferred to the E. faecalis transconjugant of the 1207/14 isolate (Table 2). Formation of larger cointegrates, likely with an involvement of very ubiquitous IS1216, might explain the existence of larger, approximately 170 kb plasmids hybridizing with the vanA probe, apart from the main 21.6 kb form of the vanA-plasmid. Formation of such fusions was indeed demonstrated for larger conjugative plasmids and smaller mobilizable poxtA-plasmids, enabling transfer of linezolid resistance from E. faecalis and Enterococcus lactis [62]. The majority of repUS1pVEF1-vanA plasmids represented multireplicons, which contained also rep18a, located upstream Tn1546 and in some instances repUS1pVEF1-vanA plasmids carried also rep1, rep2 and/or rep9. Multireplicons with various combinations of rep genes were present also in our plasmid reference set and in some instances such plasmids might represent more stable fusions formed during conjugation, described above. However, it is worth emphasizing that the presence of plasmids of very similar sizes, indistinguishable by PFGE-S1, may provide an alternative explanation for detecting multireplicons by hybridization in some isolates.
In conclusion, in Poland VanA-E. faecalis represents a heterogonous group of isolates that emerged from the general population of this species through several acquisitions of vanA determinants. The broad-host range mobilizable repUS1pVEF1-Inc18 replicons were the most common carriers of Tn1546 and these plasmids played an important role in the dissemination of VanA glycopeptide resistance among E. faecalis. Association of vanA with specific genetic background, i.e. mostly with multiresistant CCs 2 and 87, contributed to efficient spread of VanA-E. faecalis in and among hospitals in Poland, constituting a significant threat to public health.
Data availability
The datasets generated during and/or analysed during the current study are available in The National Center for Biotechnology Information (NCBI) repository, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA731638. The accession numbers of genomic sequences of particular isolates are provided in Table 2.
Code availability
Not applicable.
References
European Centre for Disease Prevention and Control (2020) Antimicrobial resistance in the EU/EEA (EARS-Net) - Annual Epidemiological Report 2019. ECDC, Stockholm
Leavis HL, Bonten MJ, Willems RJ (2006) Identification of high-risk enterococcal clonal complexes: global dispersion and antibiotic resistance. Curr Opin Microbiol 9:454–460. https://doi.org/10.1016/j.mib.2006.07.001
Kawalec M, Pietras Z, Danilowicz E, Jakubczak A, Gniadkowski M, Hryniewicz W, Willems RJ (2007) Clonal structure of Enterococcus faecalis isolated from Polish hospitals: characterization of epidemic clones. J Clin Microbiol 45:147–153. https://doi.org/10.1128/JCM.01704-06
McBride SM, Fischetti VA, Leblanc DJ, Moellering RC Jr, Gilmore MS (2007) Genetic diversity among Enterococcus faecalis. PLoS One 2:e582. https://doi.org/10.1371/journal.pone.0000582
Kuch A, Willems RJ, Werner G, Coque TM, Hammerum AM, Sundsfjord A, Klare I, Ruiz-Garbajosa P, Simonsen GS, van Luit-Asbroek M, Hryniewicz W, Sadowy E (2012) Insight into antimicrobial susceptibility and population structure of contemporary human Enterococcus faecalis isolates from Europe. J Antimicrob Chemother 67:551–558. https://doi.org/10.1093/jac/dkr544
Pöntinen AK, Top J, Arredondo-Alonso S, Tonkin-Hill G, Freitas AR, Novais C, Gladstone RA, Pesonen M, Meneses R, Pesonen H, Lees JA, Jamrozy D, Bentley SD, Lanza VF, Torres C, Peixe L, Coque TM, Parkhill J, Schürch AC, Willems RJL, Corander J (2021) Apparent nosocomial adaptation of Enterococcus faecalis predates the modern hospital era. Nat Commun 12:1523. https://doi.org/10.1038/s41467-021-21749-5
Hollenbeck BL, Rice LB (2012) Intrinsic and acquired resistance mechanisms in enterococcus. Virulence 3:421–433. https://doi.org/10.4161/viru.21282
Ayobami O, Willrich N, Reuss A, Eckmanns T, Markwar R (2020) The ongoing challenge of vancomycin-resistant Enterococcus faecium and Enterococcus faecalis in Europe: an epidemiological analysis of bloodstream infections. Emerg Microbes Infect 9:1180–1193. https://doi.org/10.1080/22221751.2020.1769500
Noble WC, Virani Z, Cree RG (1992) Co-transfer of vancomycin and other resistance genes from Enterococcus faecalis NCTC 12201 to Staphylococcus aureus. FEMS Microbiol Lett 72:195–198. https://doi.org/10.1016/0378-1097(92)90528-v
Zhu W, Murray PR, Huskins WC, Jernigan JA, McDonald LC, Clark NC, Anderson KF, McDougal LK, Hageman JC, Olsen-Rasmussen M, Frace M, Alangaden GJ, Chenoweth C, Zervos MJ, Robinson-Dunn B, Schreckenberger PC, Reller LB, Rudrik JT, Patel JB (2010) Dissemination of an Enterococcus Inc18-like vanA plasmid associated with vancomycin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 54:4314–4320. https://doi.org/10.1128/AAC.00185-10
Arthur M, Molinas C, Depardieu F, Courvalin P (1993) Characterization of Tn1546, a Tn3-related transposon conferring glycopeptide resistance by synthesis of depsipeptide peptidoglycan precursors in Enterococcus faecium BM4147. J Bacteriol 175:117–127. https://doi.org/10.1128/jb.175.1.117-127.1993
Dahl KH, Lundblad EW, Røkenes TP, Olsvik Ø, Arnfinn Sundsfjordet A (2000) Genetic linkage of the vanB2 gene cluster to Tn5382 in vancomycin-resistant enterococci and characterization of two novel insertion sequences. Microbiology 146:1469–1479. https://doi.org/10.1099/00221287-146-6-1469
Garnier F, Taourit S, Glaser P, Courvalin P, Galimand M (2000) Characterization of transposon Tn1549, conferring VanB-type resistance in Enterococcus spp. Microbiology 146:1481–1489. https://doi.org/10.1099/00221287-146-6-1481
Kawalec M, Gniadkowski M, Hryniewicz W (2000) Outbreak of vancomycin-resistant enterococci in a hospital in Gdansk, Poland, due to horizontal transfer of different Tn1546-like transposon variants and clonal spread of several strains. J Clin Microbiol 38:3317–3322. https://doi.org/10.1128/JCM.38.9.3317-3322.2000
Freitas AR, Novais C, Tedim AP, Francia MV, Baquero F, Peixe L, Coque TM (2013) Microevolutionary events involving narrow host plasmids influences local fixation of vancomycin-resistance in Enterococcus populations. PLoS One 8:e60589. https://doi.org/10.1371/journal.pone.0060589
Rosvoll TC, Pedersen T, Sletvold H, Johnsen PJ, Sollid JE, Simonsen GS, Jensen LB, Nielsen KM, Sundsfjord A (2010) PCR-based plasmid typing in Enterococcus faecium strains reveals widely distributed pRE25-, pRUM-, pIP501- and pHTbeta-related replicons associated with glycopeptide resistance and stabilizing toxin-antitoxin systems. FEMS Immunol Med Microbiol 58:254–268. https://doi.org/10.1111/j.1574-695X.2009.00633.x
Raven KE, Reuter S, Gouliouris T, Reynolds R, Russel JE, Brown NM, Török ME, Parkhill J, Peacock SJ (2016) Genome-based characterization of hospital-adapted Enterococcus faecalis lineages. Nat Microbiol 1:15033. https://doi.org/10.1038/nmicrobiol.2015.33
Wardal E, Gawryszewska I, Hryniewicz W, Sadowy E (2013) Abundance and diversity of plasmid-associated genes among clinical isolates of Enterococcus faecalis. Plasmid 70:329–342. https://doi.org/10.1016/j.plasmid.2013.07.003
Wardal E, Sadowy E (2021) Complete genome sequence of a Polish Enterococcus faecalis vanA-positive hospital isolate. Microbiol Resour Announc 10:e00668-e721. https://doi.org/10.1128/MRA.00668-21
Buetti N, Wassilew N, Rion V, Senn L, Gardiol C, Widmer A, Marschall J; for Swissnoso. Emergence of vancomycin-resistant enterococci in Switzerland: a nation-wide survey. Antimicrob Resist Infect Control 17:16. https://doi.org/10.1186/s13756-019-0466-x
The European Committee on Antimicrobial Susceptibility Testing (2021) Breakpoint tables for interpretation of MICs and zone diameters. Version 11:2021
Lanyi B (1987) Classical and rapid identification methods for medically important bacteria. Methods Microbiol 19:1–67. https://doi.org/10.1016/S0580-9517(08)70407-0
Franz CM, Muscholl-Silberhorn AB, Yousif NM, Vancanneyt M, Swings J, Holzapfel WH (2001) Incidence of virulence factors and antibiotic resistance among enterococci isolated from food. Appl Environ Microbiol 67:4385–4389. https://doi.org/10.1128/aem.67.9.4385-4389.2001
Clark NC, Cooksey RC, Hill BC, Swenson JM, Tenover FC (1993) Characterization of glycopeptide-resistant enterococci from U.S. hospitals. Antimicrob Agents Chemother 37:2311–2317. https://doi.org/10.1128/aac.37.11.2311
Dahl KH, Simonsen GS, Olsvik Ø, Sundsfjord A (1999) Heterogeneity in the vanB gene cluster of genomically diverse clinical strains of vancomycin-resistant enterococci. Antimicrob Agents Chemother 43:1105–1110. https://doi.org/10.1128/AAC.43.5.1105
Ruiz-Garbajosa P, Bonten MJ, Robinson DA, Top J, Nallapareddy SR, Torres C, Coque TM, Canton R, Baquero F, Murray BE, del Campo R, Willems RJ (2006) Multilocus sequence typing scheme for Enterococcus faecalis reveals hospital-adapted genetic complexes in a background of high rates of recombination. J Clin Microbiol 44:2220–2228. https://doi.org/10.1128/JCM.02596-05
Jolley KA, Bray JE, Maiden MCJ (2018) Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res 24:124. https://doi.org/10.12688/wellcomeopenres.14826.1
Gawryszewska I, Żabicka D, Hryniewicz W, Sadowy E (2021) Penicillin-resistant, ampicillin-susceptible Enterococcus faecalis in Polish hospitals. 27:291–300. https://doi.org/10.1089/mdr.2019.0504
de Lencastre H, Severina EP, Roberts RB, Kreiswirth BN, Tomasz A, The BARG Initiative Pilot Study Group. Bacterial Antibiotic Resistance Group (1996) Testing the efficacy of a molecular surveillance network: methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus faecium (VREF) genotypes in six hospitals in the metropolitan New York City area. Microb Drug Resist 2:343–351. https://doi.org/10.1089/mdr.1996.2.343
Clark NC, Cooksey RC, Hill BC, Swenson JM, Tenover FC (1993) Characterization of glycopeptide-resistant enterococci from U.S. hospitals. Antimicrobl Agents Chemother 37:2311–2317. https://doi.org/10.1128/aac.37.11.2311
Wardal E, Kuch A, Gawryszewska I, Zabicka D, Hryniewicz W, Sadowy E (2017) Diversity of plasmids and Tn1546-type transposons among VanA Enterococcus faecium in Poland. Eur J Clin Microbiol Infect Dis 36:313–328. https://doi.org/10.1007/s10096-016-2804-8
Jensen LB, Garcia-Migura L, Valenzuela AJ, Lohr M, Hasman H, Aarestrup FM (2010) A classification system for plasmids from enterococci and other Gram-positive bacteria. J Microbiol Methods 80:25–43. https://doi.org/10.1016/j.mimet.2009.10.012
Barton BM, Harding GP, Zuccarelli AJ (1995) A general method for detecting and sizing large plasmids. Anal Biochem 226:235–240. https://doi.org/10.1006/abio.1995.1220
Manson JM, Hancock LE, Gilmore MS (2010) Mechanism of chromosomal transfer of Enterococcus faecalis pathogenicity island, capsule, antimicrobial resistance, and other traits. Proc Natl Acad Sci USA 107:12269–12274. https://doi.org/10.1073/pnas.1000139107
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:12. https://doi.org/10.14806/ej.17.1.200
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. https://doi.org/10.1089/cmb.2012.0021
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30(14):2068–2069. https://doi.org/10.1093/bioinformatics/btu153
Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, Fookes M, Falush D, Keane JA, Parkhill J (2015) Roary: rapid large-scale prokaryote pangenome analysis. Bioinformatics 31:3691–3693. https://doi.org/10.1093/bioinformatics/btv421
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Carattoli A, Zankari E, Garcia-Fernandez A, Voldby Larsen M, Lund O, Villa L, Moller Aarestrup F, Hasman H (2014) In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother 58:3895–3903. https://doi.org/10.1128/AAC.02412-14
Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV 67 (2012) Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67:2640–2644. https://doi.org/10.1093/jac/dks261
Li X, Xie Y, Liu M, Tai C, Sun J, Deng Z, Ou H-Y (2018) oriTfinder: a web-based tool for the identification of origin of transfers in DNA sequences of bacterial mobile genetic elements. Nucleic Acids Res 46(W1):W229–W234. https://doi.org/10.1093/nar/gky352
Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA (2011) BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. https://doi.org/10.1186/1471-2164-12-402
Wardal E, Markowska K, Zabicka D, Wroblewska M, Giemza M, Mik E, Polowniak-Pracka H, Wozniak A, Hryniewicz W, Sadowy E (2014) Molecular analysis of vanA outbreak of Enterococcus faecium in two Warsaw hospitals: the importance of mobile genetic elements. Biomed Res Int 2014:575367. https://doi.org/10.1155/2014/575367
Willems RJ, Top J, van den Braak N, van Belkum A, Mevius DJ, Hendriks G, van Santen-Verheuvel M, van Embden JD (1999) Molecular diversity and evolutionary relationships of Tn1546-like elements in enterococci from humans and animals. Antimicrob Agents Chemother 43:483–491. https://doi.org/10.1128/AAC.43.3.483
Jensen LB, Ahrens P, Dons L, Jones RN, Hammerum AM, Aarestrup FM (1998) Molecular analysis of Tn1546 in Enterococcus faecium isolated from animals and humans. J Clin Microbiol 36:437–442. https://doi.org/10.1128/JCM.36.2.437-442.1998
Weaver KE, Jensen KD, Colwell A, Sriram SI (1996) Functional analysis of the Enterococcus faecalis plasmid pAD1-encoded stability determinant par. Mol Microbiol 20:53–63. https://doi.org/10.1111/j.1365-2958.1996.tb02488.x
Garcillán-Barcia MP, Francia MV, de la Cruz F (2009) The diversity of conjugative relaxases and its application in plasmid classification. FEMS Microbiol Rev 33:657–687. https://doi.org/10.1111/j.1574-6976.2009.00168.x
Chen Y, Staddon JH, Dunny GM (2007) Specificity determinants of conjugative DNA processing in the Enterococcus faecalis plasmid pCF10 and the Lactococcus lactis plasmid pRS01. Mol Microbiol 63:1549–1564. https://doi.org/10.1111/j.1365-2958.2007.05610.x
Francia MV, Clewell DB (2002) Amplification of the tetracycline resistance determinant of pAMalpha1 in Enterococcus faecalis requires a site-specific recombination event involving relaxase. J Bacteriol 184:5187–5193. https://doi.org/10.1128/JB.184.18.5187-5193.2002
Sletvold H, Johnsen PJ, Simonsen GS, Aasnaes B, Sundsfjord A, Nielsen KM (2007) Comparative DNA analysis of two vanA plasmids from Enterococcus faecium strains isolated from poultry and a poultry farmer in Norway. Antimicrob Agents Chemother 51:736–739. https://doi.org/10.1128/AAC.00557-06
Inoue T, Tomita H, Ike Y (2006) Bac 32, a novel bacteriocin widely disseminated among clinical isolates of Enterococcus faecium. Antimicrob Agents Chemother 50:1202–1212. https://doi.org/10.1128/AAC.50.4.1202-1212.2006
Bashir A, Attie O, Sullivan M, Sebra R, Singh KV, Altman D, Pak T, Dutta J, Chacko K, Webster E, Lewis M, Hamula C, Delli Carpini KW, Murray BE, Kasarskis A, van Bakel H, Huprikar S (2017) Genomic confirmation of vancomycin-resistant Enterococcus transmission from deceased donor to liver transplant recipient. PLoS One 12:e0170449. https://doi.org/10.1371/journal.pone.0170449.eCollection2017
Wang G, Yu F, Lin H, Murugesan K, Huang W, Hoss AG, Dhand A, Lee LY, Zhuge J, Yin C, Montecalvo M, Dimitrova N, Fallon JT (2018) Evolution and mutations predisposing to daptomycin resistance in vancomycin-resistant Enterococcus faecium ST736 strains. PLoS One 13:e0209785. https://doi.org/10.1371/journal.pone.0209785
Biggel M, Nüesch-Inderbinen M, Raschle S, Stevens MJA, Stephan R (2021) Spread of vancomycin-resistant Enterococcus faecium ST133 in the aquatic environment in Switzerland. J Glob Antimicrob Resist 27:31–36. https://doi.org/10.1016/j.jgar.2021.08.002
Rao C, Dhawan B, Vishnubhatla S, Kapil A, Das B, Sood S (2020) Emergence of high-risk multidrug-resistant Enterococcus faecalis CC2 (ST181) and CC87 (ST28) causing healthcare-associated infections in India. Infect Genet Evol 85:104519. https://doi.org/10.1016/j.meegid.2020.104519
Woodford N, Adebiyi AM, Palepou MF, Cookson BD (1998) Diversity of VanA glycopeptide resistance elements in enterococci from humans and nonhuman sources. Antimicrob Agents Chemother 42:502–508. https://doi.org/10.1128/AAC.42.3.502
Lim SK, Tanimoto K, Tomita H, Ike Y (2006) Pheromone-responsive conjugative vancomycin resistance plasmids in Enterococcus faecalis isolates from humans and chicken feces. Appl Environ Microbiol 72:6544–6553. https://doi.org/10.1128/AEM.00749-06
Sletvold H, Johnsen PJ, Wikmark OG, Simonsen GS, Sundsfjord A, Nielsen KM (2010) Tn1546 is part of a larger plasmid-encoded genetic unit horizontally disseminated among clonal Enterococcus faecium lineages. J Antimicrob Chemother 65:1894–1906. https://doi.org/10.1093/jac/dkq219
Freitas AR, Coque TM, Novais C, Hammerum AM, Lester CH, Zervos MJ, Donabedian S, Jensen LB, Francia MV, Baquero F, Peixe L (2011) Human and swine hosts share vancomycin-resistant Enterococcus faecium CC17 and CC5 and Enterococcus faecalis CC2 clonal clusters harboring Tn1546 on indistinguishable plasmids. J Clin Microbiol 49:925–931. https://doi.org/10.1128/JCM.01750-10
Laverde Gomez JA, Hendrickx AP, Willems RJ, Top J, Sava I, Huebner J, Witte W, Werner G (2011) Intra- and interspecies genomic transfer of the Enterococcus faecalis pathogenicity island. PLoS One 6:e16720. https://doi.org/10.1371/journal.pone.0016720
Shan X, Yang M, Wang N, Schwarz S, Li D, Du XD (2022) Plasmid fusion and recombination events that occurred during conjugation of poxtA-carrying plasmids in enterococci. Microbiol Spectr 10:e0150521. https://doi.org/10.1128/spectrum.01505-21
Acknowledgements
We thank all Polish microbiologists who have submitted isolates for this study.
Funding
This work was supported by the grant UMO-2014/15/N/NZ7/02960 from the National Science Centre (NCN, Poland). Isolate collection and maintenance was supported by the grant Narodowy Program Ochrony Antybiotyków (NPOA) from the Ministry of Health and by the grant SPUB MIKROBANK from the Ministry of Science and Higher Education. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
The isolates used in the current study were obtained during a routine national surveillance activity of the National Reference Centre for Susceptibility Testing, under the mandate of the Ministry of Health. The study was performed in a retrospective manner with an anonymization of patients’ data; thus, ethical approval and informed consent were not required.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
10096_2022_4479_MOESM1_ESM.pptx
Supplementary file 1 Supplementary Fig. 1. Genome-based relationships among CC87 (A) and ST6 (B) isolates from Poland and other countries. The ML tree (left side of the figure) was constructed with RAxML based on core genome alignment, generated in Roary. Twenty-three genomic sequences of isolates belonging to CC87 and 33 representatives of ST6 were downloaded from GenBank (23rd February 2022) and supplemented with available data on isolation country, source and year (right side of the figure). Two-letter country code follows the international standard ISO3166-1 alpha-2 (https://www.iso.org/iso-3166-country-codes.html; 22nd March 2022 date last accessed). Y, presence; 0, absence; n/a, data not available; data for isolates from the current study in bold. In the tree constructed for ST6 (B), isolates represented by a single representative isolate (underlined) are provided within brackets as follows: 1248_EFLS (1308_EFLS), 132_EFLS (133_EFLS), 1333_EFLS (1325_EFLS), V583 (V587, NCTC13379), B1586 (B1005, B1290, B1376, B4148, B878, B939, B1327, B1696, B1719, B2593, B2867, B3053, B4267, B4259, B4568, B4672, B4411, B1851, B1138, B5076, B1249, B1843, B2391, B2813, B3126, B4008, B4674, B2670, B4018, B5035, B1623, B1933, B2535, B2557, B1532, B1734, B2949, B1505, B1678, B1874, B2202, B2255, B2211, B2277, B2488, B2687, B2685, B2864, B3042, B3286, B4163, B4638, B4969, B3031, B3119, B3196, B2802, EnGen0427), SF100 (SF19), VRE32631 (VRE33430, VRE33236, VRE33319, VRE33481, VRE34517, VRE34684, VRE33143, VRE33492, VRE33271, VRE33454, VRE32839, VRE33670), VRE32867 (VRE32870), VRE32924 (VRE32954, VRE33211), VRE32879 (VRE33353, VRE33801, VRE32930, VRE33535, VRE34808, VRE33107, VRE33257, VRE33766).(PPTX 389 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wardal, E., Żabicka, D., Hryniewicz, W. et al. VanA-Enterococcus faecalis in Poland: hospital population clonal structure and vanA mobilome. Eur J Clin Microbiol Infect Dis 41, 1245–1261 (2022). https://doi.org/10.1007/s10096-022-04479-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-022-04479-4