
Abstract
We described the population structure of Bordetella pertussis (B. pertussis) in Norway from 1996 to 2019 and determined if there were evolutionary shifts and whether these correlated with changes in the childhood immunization program. We selected 180 B. pertussis isolates, 22 from the whole cell vaccine (WCV) era (1996–1997) and 158 from the acellular vaccine (ACV) era (1998–2019). We conducted whole genome sequencing and determined the distribution and frequency of allelic variants and temporal changes of ACV genes. Norwegian B. pertussis isolates were evenly distributed across a phylogenetic tree that included global strains. We identified seven different allelic profiles of ACV genes (A–F), in which profiles A1, A2, and B dominated (89%), all having pertussis toxin (ptxA) allele 1, pertussis toxin promoter (ptxP) allele 3, and pertactin (prn) allele 2 present. Isolates with ptxP1 and prn1 were not detected after 2007, whereas the prn2 allele likely emerged prior to 1972, and ptxP3 before the early 1980s. Allele conversions of ACV genes all occurred prior to the introduction of ACV. Sixteen percent of our isolates showed mutations within the prn gene. ACV and its booster doses (implemented for children in 2007 and adolescents in 2013) might have contributed to evolvement of a more uniform B. pertussis population, with recent circulating strains having ptxA1, ptxP3, and prn2 present, and an increasing number of prn mutations. These strains clearly deviate from ACV strains (ptxA1, ptxP1, prn1), and this could have implications for vaccine efficiency and, therefore, prevention and control of pertussis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Pertussis, caused by Bordetella pertussis (B. pertussis), is a highly contagious acute respiratory infection. The disease affects people of all ages, but is especially severe in infants. Although pertussis is a vaccine-preventable disease with high vaccine coverage worldwide, a global resurgence of this infection has been observed [1]. Pertussis is endemic in many countries, with epidemic cycles occurring every 2–5 years [2]. In 2014, 24.1 million pertussis cases were estimated globally, with 160,700 deaths from pertussis in children less than 5 years of age [3]. In Europe, a notification rate of 8.2 cases per 100,000 population was reported in 2018, with the highest notification rate in infants followed by 10–14-year olds [4]. In Norway, a notification rate of 47.4 cases per 100,000 population was reported in 2019 (www.msis.no). Norway has, since 2011, consistently reported the highest notification rate of pertussis in all of Europe, and in contrast to the majority of European countries, adults (≥ 18 years of age) accounted for the majority of cases (55%) [5].
During the mid-1990s and in the early twenty-first century, whole cell vaccines (WCVs) were replaced by acellular pertussis vaccines (ACVs) in high-income countries, including Norway, where ACVs were introduced from 1998 [2]. To combat the increasing incidence of pertussis, Norway and other European countries implemented childhood and adolescent ACV boosters [6, 7]. In Norway, ACV boosters were introduced for 7-year olds in 2006/2007 and for 15-year olds in 2013/2014. Recently, maternal immunization was introduced in some countries to prevent severe pertussis in infants, but has yet to be introduced in Norway [8].
Several factors are contributing to the re-emergence of pertussis, like improved diagnostics [9], increased awareness of the disease [10, 11], decreased vaccine efficacy [12], waning immunity [8, 13, 14], and pathogen adaption [2, 15,16,17,18,19]. Growing evidence suggests that genetic divergence of circulating B. pertussis population away from vaccine antigens and the emergence of strains with increased pertussis toxin production might be contributing factors [17, 19,20,21]. Deficiency in pertactin (Prn), one of the components in ACVs, is widely distributed globally, and the rapid spread of Prn deficiency is likely vaccine driven [22]. Mutations in other ACV antigens such as pertussis toxin (ptxA), its promoter (ptxP), and fimbria (fim2 and fim3) are also reported and shown to have quickly spread throughout the B. pertussis population [17, 23]. Currently, more than 90% of B. pertussis circulating in Europe have ptxA1, ptxP3, and prn2 genotypes and the recent replacement of ptxP1 with ptxP3 is associated with increased pertussis toxin production [17, 19, 24]. Comparative genomic analysis has demonstrated that worldwide transmission of new strains is rapid, and that the global population of B. pertussis is evolving in response to vaccine introduction, potentially enabling vaccine escape [17, 25]. All these changes raise concern, as ACV-induced selection pressure could potentially threaten vaccine efficacy [22, 26].
Pertussis has resurged in Norway with high notification rates since the late 1990s. Although lethal cases are very rare, pertussis is poorly controlled [7, 27]. No overview of the molecular epidemiological situation of B. pertussis in Norway exists. Only a few Norwegian isolates from the last decade have previously been included in European studies, showing that the ptxA1, ptxP3, and prn2 genotypes are frequent and that Prn-deficient isolates are present among isolates examined [16, 18, 20, 28]. In Norway, the three-component ACVs used in the childhood immunization program and as booster vaccine for 15-year olds include vaccine antigens from Tohama I: ptxA2, ptxP1, prn1, and fhaB1 (https://bigsdb.pasteur.fr/bordetella/), whereas the two-component vaccine used as a booster vaccine for 7-year olds harbors vaccine antigens from the “Pillemer (P134)” strain, with ptxA2 present, but no information on the allelic variant of fhaB [29]. This suggests that the Norwegian B. pertussis population might have ACV antigen profiles evolving away from the antigens used in ACVs. The aim of this retrospective study was to characterize the population structure of B. pertussis in Norway over the period from 1996 to 2019 and determine whether there have been antigenic shifts in the wake of the introduction of ACVs. As such change may have implications for vaccine efficiency, the results can inform vaccine policy in Norway.
Material and methods
B. pertussis isolates
Clinical microbiological laboratories from each of five regions of Norway (Eastern Norway, Southern Norway, Western Norway, Northern Norway, and Trøndelag County) referred B. pertussis isolates or PCR-positive samples from clinical specimens to the National Reference Laboratory (NRL) for pertussis at the Norwegian Institute of Public Health (NIPH) on a voluntary basis. Before 2002, pertussis cases were diagnosed with culture and serological methods; however, from 2002, the use of PCR gradually increased, first among the youngest age groups, and from 2012, the majority of cases with pertussis in Norway were diagnosed by PCR. Stratified convenience sampling of 180 B. pertussis isolates from the microbiological biobank at the NRL was performed, taking into account the time of sampling (year), place of residence (five regions of Norway), and age of the case at the time of sampling (< 20 years or ≥ 20 years). We included eight and 14 isolates from 1996 and 1997, the WCV era, respectively, and a median of eight (range 5–10) isolates annually from 1998 to 2019, the ACV era (Table S1). No isolates or PCR-positive samples were received at the NRL for the years 2010 and 2011. Twenty-five of the isolates have previously been included in European Pertussis Surveillance studies [16, 18, 20, 28] (Table S1).
Culture, DNA extraction, and whole genome sequencing
Approximately 60 PCR-positive clinical samples were cultured on charcoal agar with 40 µg/ml cephalexin and 250 µg/ml amphotericin B (made in-house at the NIPH) at 35℃ without CO2 under humid conditions for up to 10 days. Pure cultures were verified by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) (Daltonics) and stored in the microbiological biobank at − 70℃. Strains selected from the biobank were cultured on charcoal agar as described above for 24–72 h prior to genomic DNA extraction using QIAamp DNA Mini QIAcube kit (Qiagen) on QIAcube (Qiagen) following the manufacturer’s procedure. DNA concentration was measured using a Qubit fluorometer (Invitrogen, Thermo Fisher Scientific). DNA libraries were constructed with KAPA HyperPlus (Roche Life Science) with KAPA Unique Dual-Indexed Adapter kit (Roche Life Science), following the manufacturer’s instructions. Clean-up and size selection were performed using magnetic AMPure XP beads (Beckman Coulter). DNA was then quantified using a Qubit fluorometer, and the size of fragments was measured using an Agilent 4200 TapeStation system (Agilent Technologies). Paired-end reads (300 bp × 2) were produced on an Illumina MiSeq platform according to standard protocols using the MiSeq Reagent kits (v2 600 cycles; Illumina). Illumina data was deposited in the European Nucleotide Archive under the accession numbers ERS7736033 to ERS7736212. FastQC (Babraham Bioinformatics) was used for quality control of the raw reads. Sequence reads were trimmed and adapters removed using Trimmomatic v0.36. FASTA contigs were obtained from the trimmed reads using SPAdes v3.13.2. The final assembly files consisted of a median number of 277 contigs/sample (range: 261 to 1568 contigs) and had a median of 75.9 coverage/sample (range: 14.9 to 520.4 coverage). The mean length/sample was 3,885,772 bp. The B. pertussis genome is ~ 4.1 Mb, and the estimated mean coverage across all sequencing runs was 94.2%. Kraken (version 1.1.1) was used to assign taxonomic label species identification. All, except one sample, showed pure cultures of B. pertussis. The last sample was contaminated with Streptococcus (Table S1).
Multilocus sequence typing
Multilocus sequence typing (MLST) according to the Institut Pasteur scheme (https://bigsdb.pasteur.fr/bordetella/) [30] was obtained using the program mlst v2.15 (https://github.com/tseemann/mlst) on the filtered FASTA contigs described above.
Allelic variants of vaccine antigen genes and presence of molecular markers of erythromycin resistance
Sequences of allele variants of genes encoding vaccine antigens: pertussis toxin (ptxA, 39 alleles), pertussis toxin promoter (ptxP, 40 alleles), pertactin (prn, 140 alleles), fimbriae 2 (fim2, 15 alleles), fimbriae 3 (fim3, 38 alleles), and filamentous hemagglutinin B (fhaB, 100 alleles) were downloaded from the Institut Pasteur database at https://bigsdb.pasteur.fr/bordetella/. For assembled genomes, in silico typing of the vaccine antigen genes and screening for 23S ribosomal RNA (rRNA) A2037G (in Tohama I, NC_002929.2) mutation and the presence of erythromycin-resistant genes in the ResFinder database (2021–04-20) were performed using BLAST + tools (version 2.6.0) with strict criteria (100% match). Based on sequence similarities in Pasteur prn alleles 3 and 108, i.e., missing a 15-bp insertion (GGT CCC GGC GGC TTC at position 864) or having one single nucleotide polymorphism (SNP) (C > G at position 763) compared to prn2, respectively, these were interpreted as prn2 to be comparable with previous publications (Table S1). Similarly, Pasteur prn allele 55, with one SNP (C > T) at position 2526 compared to prn1, was interpreted as prn1.
Defining molecular mechanisms associated with mutations within the prn gene
A region including the prn gene was extracted from all 180 whole genome sequences (approximately 2748 bp). The sequences were imported and aligned in MEGA X [31] and manually examined for mutations using Tohama I (NC_002929.2), carrying prn1, as the reference. All sequences were screened for insertion sequence (IS) elements in prn using ISMapper (https://github.com/jhawkey/IS_mapper).
Global B. pertussis sequence collection
Sequencing data of 371 publicly available B. pertussis strains isolated from cases identified in six different continents, Asia (n = 12), Africa (n = 2), North America (n = 300), South America (n = 8), Europe (n = 43), and Oceania (n = 5), were included in our study [32] (Table S2). The sequences were obtained both from long-sequence technology (PacBio) and short-sequence technology (Illumina). Complete genomes were available for 319 (86%) of the strains.
SNP identification and phylogenetic analysis comparing the Norwegian B. pertussis population with the global population
After considering 319 publicly available closed B. pertussis genomes (Table S2), B1917 (accession: NZ_CP009751.1) was chosen as the most appropriate reference genome in the SNP analysis, having the highest degree of genomic homology to the Norwegian isolates included in this study, as assessed with HarvestTools v1.2 (http://dx.doi.org/10.1186/s13059-014-0524-x). Notably, Tohama I, the vaccine strain, was more distantly related to the Norwegian isolates than nearly every other publicly available B. pertussis genomes. In order to identify SNPs, reads were mapped to the B1917 genome using the Snippy pipeline v4.6.0 (https://github.com/tseemann/snippy). Maximum likelihood phylogenetic tree was created using iqtree v2.1.2 (https://doi.org/10.1093/molbev/msu300; https://doi.org/10.1038/nmeth.4285) with automatic model selection.
Time-measured evolutionary analysis
Phylogenetic tree scaled to time was created using the program treetime v0.8.1 with the following non-default options: least-squares rerooting, accounting for covariation, and coalescent rate set to constant. The R2 value was 0.63. Figures were annotated using ITOL [33].
Results
Allelic profiles of ACV gene variants and the presence of molecular markers of erythromycin resistance
The majority of the Norwegian B. pertussis isolates showed sequence type (ST) 2 (96%, 172/180), whereas the remaining had ST83. All our isolates carried ptxA1. The ptxP3 allele was dominating in our isolates (88%, 159/180), even though in 1996 and 1997, prior to introduction of ACV in Norway, ptxP1 was present in 63% (5/8) and 71% (10/14) of the selected isolates, respectively (Fig. 1). Ninety-eight percent (177/180) of our isolates harbored prn2, and they showed Pasteur prn allele 2 (prn2, 159/177), allele 108 (16/177), and allele 3 (2/177) (Table 1 and Table S1). The three remaining isolates (2%, 3/180) carried prn1 (one of the isolates showed Pasteur prn allele 55). The majority of our B. pertussis isolates had the fim2-1 allele present (99%, 178/180). One isolate harbored the fim2-2 allele, whereas the remaining isolate carried fim2-15 (Table S1). The fim3-1 allele was present in 52% (94/180) of our isolates, whereas 47% (85/180) had fim3-2. The remaining isolate carried fim3-4 (Table S1). The fim3-1 allele was dominating prior to introduction of ACV in Norway in 1998 but was also common from 2007 and forward (Fig. 1). The majority of the Norwegian B. pertussis isolates carried fhaB1 (85%, 153/180), whereas 15% (27/180) had the fhaB7 allele (G > A at position 948 compared to fhaB1) (Table S1 and Fig. 1).

Distribution of allelic variants of the genes encoding acellular vaccine (ACV) antigens in the Norwegian B. pertussis population (n = 180), 1996–2019. The alleles ptxA1, ptxP3, prn2, fim2-1, and fhaB1 were dominating in our isolates, whereas a more even distribution of the fim3-1 and fim3-2 alleles was seen. ptxP1 was more common prior to introduction of ACV, whereas ptxP3 dominated thereafter. Whole cell vaccine (WCV) period: 1996 and 1997; ACV period: 1998–2019. For the years 2010 and 2011, no B. pertussis isolates or PCR-positive samples were received at the National Reference Laboratory for pertussis at the Norwegian Institute of Public Health. Alleles were assigned using the following database: https://bigsdb.pasteur.fr/bordetella/ (see text for details)
Seven different allelic profiles were identified (profiles A1–F, Table 2). Profile A1 (ptxA1, ptxP3, prn2, fim2-1, fim3-2, fhaB1) and profile A2: identical to A1, except for replacement of fhaB1 with fhaB7, occurred in 32% (58/180) and 15% (27/180) of the Norwegian B. pertussis isolates, respectively. Profile B (ptxA1, ptxP3, prn2, fim2-1, fim3-1, and fhaB1) was present in 42% (75/180), including one isolate with ptxP34 instead of ptxP3 and another isolate with fim2-15 instead of fim2-1 (both indicated as profile B* in Table S1). Profile C (ptxA1, ptxP1, prn2, fim2-1, fim3-1, and fhaB1) was seen in 9.4% (17/180) of the isolates. Two of the isolates had prn3 instead of prn2 (indicated as profile C* in Table S1). Profiles D–F were present in one isolate each, all carrying ptxA1, ptxP1, prn1, and fhaB1. These profiles had fim2-1 or fim2-2 and fim3-1 or fim3-4 (Table 2 and Table S1). Profile C was dominating prior to ACV implementation, whereas profile A2 was most common between 1998 and 2003 (Fig. S1). Profile A2 became rarer from around 2003 and was completely eradicated from the population by the time the ACV booster dose for children was introduced in 2006/2007. The period 2003–2007 also saw the relative growth of profile A1. From 2007, coinciding introduction of the second ACV booster dose for 15-year olds in 2013/2014, profile B was dominating in the Norwegian B. pertussis population (Fig. S1).
None of the 180 B. pertussis isolates from Norway harbored mutations in the 23S rRNA gene but had wild type (A) at the SNP position A2037G (Tohama I, NC_002929.2). A single isolate from 2019 (allelic profile B) carried msr(D), an ABC transporter involved in macrolide and streptogramin B resistance, and mef(A), a marker for erythromycin and azithromycin resistance, respectively (Table S1).
Molecular mechanisms involved in mutations within the prn gene
Sixteen percent (29/180) of the Norwegian B. pertussis isolates harbored mutations within the prn gene, including partial deletions and IS481 insertions (Table 1). Among these, 97% (28/29) carried prn2 and one had prn1. Of the isolates with prn mutations, allelic profile B was dominating (79%, 23/29), whereas profile A1 occurred in four isolates (14%, 4/29) and profiles A2 and F in one isolate each, respectively. Twelve (41%) isolates had insertion of IS481 in positions between 1613 and 1614 or between 241 and 242, including isolates with allelic profiles B (n = 7), A1 (n = 4), and F (n = 1). Eight (28%) isolates, all allelic profile B, had C > T mutation leading to stop codon at positions 1258 (n = 6) and 223 (n = 2), respectively. The last nine isolates (31%) had deletion in the start of prn, eight with allelic profile B and one with profile A2. Seven of the nine isolates had deletion of IS1663 downstream of the prn gene including deletion of the proximal part of prn (Table 1). Mutations within the prn gene were more common from 2007, and in 2019, 60% (6/10) of the selected Norwegian B. pertussis isolates had prn mutations (Fig. S2).
Evolution of B. pertussis in Norway from 1996 to 2019 and its correlation to alterations in pertussis vaccination
Temporal changes of genes encoding ACV-related antigens showed that alterations of these genes occurred prior to introduction of ACV in 1998, but mainly after implementation of WCV in 1952 (Fig. 2). The prn2 allele likely emerged prior to 1972 and the ptxP3 allele before the early 1980s. B. pertussis isolates with allelic profile B (ptxA1, ptxP3, prn2, fim2-1, fim3-1, and fhaB1) were introduced around 1980, and allelic profile B is still the dominating profile of Norwegian B. pertussis isolates. B. pertussis isolates with allelic profiles C–F, all carrying ptxP1 and prn1/prn2, were present before and during the WCV era, but disappeared around 2007. In the mid-1990s, the fim3-2 allele appeared and allelic profile A1 (ptxA1, ptxP3, prn2, fim2-1, fim3-2, fhaB1) was introduced. After the mid-1990s, but before the introduction of ACV, allelic profile A2 occurred, carrying the fhaB7 allele. However, profile A2 disappeared around 2005 prior to introduction of the first ACV booster dose. After 2005, only two allelic profiles (A1 and B) were present in the Norwegian B. pertussis population, both carrying ptxA1, ptxP3, prn2, fim2-1, and fhaB1 and either fim3-2 (A1) or fim3-1 (B) (Fig. 2). An increase in the number of mutations within the prn gene was seen after the introduction of ACV and its booster doses, and these mutations were mainly seen in B. pertussis isolates with allelic profiles A1 and B (Table 1 and Fig. 2).

Molecular clock phylogeny of Norwegian B. pertussis isolates (n = 180), 1996–2019. Seven different allelic profiles of genes encoding acellular vaccine (ACV) antigens are indicated with different colors (A1, blue; A2, orange; B, red; C, turquoise; D, green; E, yellow; F, purple). Alterations of allele variants of the ACV antigens are shown as symbols (star, prn1 → prn2; right-pointing triangle, ptxP1 → ptxP3; circle, fim3-1 → fim3-2; and black check mark, fhaB1 → fhaB7). B. pertussis isolates with mutations within the prn gene compared to the prn1 allele of Tohama I (NZ_002929.2) are indicated on the figure. The red vertical lines indicate implementation of the whole cell vaccine in Norway in 1952, the ACV in 1998, the first booster dose for 7-year olds in 2006, and the second booster dose for 15-year olds in 2013. The WCV era is indicated in pink color, whereas the ACV era is indicated in green. Antigen profile B* includes one isolate with ptxP34 instead of ptxP3 and another isolate with fim2-15 instead of fim2-1. Antigen profile C* includes two isolates with prn3 instead of prn1
Comparison of the Norwegian B. pertussis population with the global population
The available global B. pertussis isolates covered the time period 1936 to 2016 and 18 countries in six different continents, although 81% (300/370) of the isolates were from North America (Table S2). The majority of the isolates showed ST2.
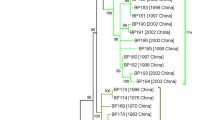
The maximum likelihood tree showed that the Norwegian B. pertussis population was distributed across the global population of ST2 and ST83 isolates (Fig. 3). However, a few branches consisted entirely of Norwegian B. pertussis isolates. Branch 1 included isolates from 1997 to 2005, and they showed allelic profile A2, with the fhaB7 allele present. Branch 2 consisted of a subgroup of Norwegian B. pertussis isolates with allelic profile B. These isolates were from 2012 to 2018, and the majority was from cases living in Eastern Norway. Branch 3 included B. pertussis from 1996 to 1998 carrying profile C, whereas branches 4 (1997–2006) and 5 (2009–2019) both consisted of isolates with allelic profile A1 (Fig. 3).

SNP-based phylogenetic tree of Norwegian B. pertussis isolates, 1996–2019, put in a global context. Norwegian strains (n = 180, purple circles) were distributed across the global phylogenetic tree; however, a few country-specific branches were detected (1–5). Worldwide strains (n = 370) were isolated from 1939 to 2016 and covered six different continents (Table S2)
Discussion
A global resurgence of pertussis has been seen after the introduction of ACVs in the 1990s [34, 35]. Changes in the B. pertussis population and pathogen adaptation have been suggested as one of the causes of this increase [2, 15, 17,18,19]. For Norway, little data was available on the B. pertussis population before this study and none of the isolates had previously been whole genome sequenced. The 180 whole genome–sequenced B. pertussis strains, isolated from 1996 to 2019, indicated clear molecular epidemiological shifts in the Norwegian population and that mutations in ACV antigens have occurred prior to and during the WCV (1952–1998) era. Our results further showed that ACV, introduced in 1998, and its two booster doses, implemented in 2006/2007 (7-year olds) and 2013/2014 (15-year olds), respectively, have led to a more uniform B. pertussis population, with two dominating allelic profiles (A1 and B) in recent years. Both profiles have the non-ACV alleles ptxA1, ptxP3, and prn2 fixed and an increasing number of mutations within the prn gene.
In the Norwegian B. pertussis population, a heterogeneous mix of strains with a distinct time-dependent shift in allelic profiles was observed. The Norwegian strains were distributed across the global phylogenetic tree, supporting previous observations of rapid strain flow of B. pertussis between countries, even though a few country-specific branches were observed [17, 21, 25, 36, 37].
Temporal changes of genes encoding the ACV antigens in the Norwegian B. pertussis population were comparable with findings from other countries. The changes supported the evidence that genetic alterations are driven by the selective pressure imposed by vaccine-induced immunity, mainly from the WCV era, and accentuated in the ACV era [15, 17, 23, 38]. In concordance with the global population, B. pertussis with ptxP3, a gene variant leading to increased pertussis toxin production and associated with more severe disease in young infants, emerged prior to the early 1980s and dominated by the end of the 1990s [15, 17, 24, 36, 39]. The prn2 allele emerged from the 1970s, as indicated by others, and is now the dominating prn allele in both WCVs and ACVs using countries, except for China and India [17, 19, 40,41,42]. Furthermore, the fim3-2 allele appeared in the start of the 1980s and has since been present alongside fim3-1 in the B. pertussis population worldwide [15, 18, 43].
Like other countries, we observed an increasing trend of mutations within the prn gene, including partial deletions and IS481 insertions, associated with time since implementing ACVs [16, 44, 45]. Even though Prn expression was not examined in our study, five of our isolates with prn mutations have previously been shown to be Prn-deficient [28]. Interestingly, few prn-negative isolates have been reported from countries still using WCVs as well as in countries without a Prn component included in their ACVs, indicating that this alteration is driven by ACVs [16, 19, 41, 42, 46,47,48]. This has clearly been demonstrated by Japan who implemented ACVs as early as 1981 followed by increased proportion of Prn-deficient strains from the mid-1990s. However, in 2012, Japan introduced ACVs without Prn and a decline in Prn-deficient strains was observed [19, 37, 47, 48].
During the WCV era, a number of antigenic allele conversions emerged, and the introduction of ACVs led to clonal expansions of strains with mutations that did not match the ACV profile. It has been shown that B. pertussis isolates with ptxP3 and Prn expressed had increased fitness during the WCV era, whereas after implementing ACV, enhanced fitness was associated with Prn-deficiency, ptxP3 and fim3-1 [37]. Consequently, the diversity in allelic profiles decreased during the ACV era and approximately 90% of recent circulating B. pertussis isolates harbor ptxA1, ptxP3, and prn2 with an increasing number of mutations within the prn gene [2, 21]. This demonstrates that the current B. pertussis population clearly deviates from the vaccine strains (ptxA2, ptxP1, prn1). This might affect the preventive potential of the ACVs and thus contribute to resurgence of pertussis [12, 38]. However, resurgence of pertussis is a complex phenomenon, and several other factors might favor the rise in pertussis disease. For instance, ACVs have been shown to be inferior to WCVs by not inducing mucosal immune response leading to increased colonization and transmission of B. pertussis and by inducing earlier waning immunity [12, 49]. Thus, novel ACVs with antigen targets matching the current circulating B. pertussis population or new targets, as well as appropriate adjuvants to stimulate both antibody and cell-mediated immune response, are highly warranted [49, 50].
Macrolide-resistant isolates are only sporadically seen in Europe, the Middle East, and North and South America [2]. This is in concordance with our findings, showing molecular determinants for macrolide resistance in only one B. pertussis isolate. This isolate carried the msrD_2 and mefA_10 genes, to our knowledge, never detected in B. pertussis previously; however, this sequence was contaminated with Streptococcus. Interestingly, in China, antibiotic abuse has led to a high level of erythromycin resistance in the B. pertussis population and contributed to the spread of B. pertussis lineages harboring ptxA1, ptxP1, and prn1, clearly deviating from the dominating clone seen in Europe, even though similar vaccine regimes were implemented both places years ago [32].
Limitations of the study include that the representativeness of our material can be questioned, because the primary laboratories in Norway submit B. pertussis samples to the NRL at the NIPH only on a voluntary basis and few isolates were included annually. Nonetheless, the similarities with previous studies indicate that our selection of isolates probably is representative for the Norwegian population. Second, we used short-sequence technology which is incapable of detecting rearrangements and large duplications responsible for the genome diversity seen in B. pertussis [51,52,53]. However, studies using long-sequence technology in combination with short sequencing technology show similar results to ours when it comes to temporal changes of ACV antigens [17, 21]. Finally, we have examined alterations at the genomic level and not at the transcriptomic or proteomic levels. The two latter might also be important for vaccine efficacy. Several countries have reported B. pertussis strains that do not express ACV antigens such as Prn, FhaB, and Ptx, although Prn deficiency is the only change frequently reported in the B. pertussis population [15, 19, 38, 54, 55].
Conclusion
We observed molecular epidemiological shifts in the Norwegian B. pertussis population during the WCV and ACV eras. ACV use and its booster doses have led to lineage-specific proliferation of more successful, ACV-deviating strains, leading to a more uniform B. pertussis population. These clones have the ptxP3 and prn2 alleles fixed and are characterized by increasing incidence of mutations within the prn gene. The emergence of B. pertussis isolates with ACV antigens deviating from the vaccine antigens might have implications for vaccine efficiency and therefore prevention and control of pertussis. We recommend continued collection of B. pertussis for surveillance purposes, and to further explore the role of genetic adaptation. Developing new vaccine candidates, including allelic variants of ACV antigens present in the current circulating population, as well as exploring novel candidates, requires attention.
Data availability
The whole genome sequences generated and analyzed during the current study are available in European Nucleotide Archive (ENA) under the accession numbers ERS7736033 to ERS7736212.
References
World Health Organization (2015) Pertussis vaccines: WHO position paper. Wkly Epidemiol Rec 90(35):433–458
Barkoff AM, He Q (2019) Molecular epidemiology of Bordetella pertussis. Adv Exp Med Biol https://doi.org/10.1007/5584_2019_402
Yeung KHT, Duclos P, Nelson EAS, Hutubessy RCW (2017) An update of the global burden of pertussis in children younger than 5 years: a modelling study. Lancet Infect Dis 17(9):974–980. https://doi.org/10.1016/s1473-3099(17)30390-0
European Centre for Disease Prevention and Control (2020) Pertussis. In: ECDC. Annual Epidemiological Report for 2018. Stockholm, ECDC.
European Centre for Disease Prevention and Control (2019) Pertussis. Annual Epidemiological Report for 2017. Stockholm, ECDC
Zeddeman A, Witteveen S, Bart MJ, van Gent M, van der Heide HG, Heuvelman KJ et al (2015) Studying Bordetella pertussis populations by use of SNPeX, a simple high-throughput single nucleotide polymorphism typing method. J Clin Microbiol 53(3):838–846. https://doi.org/10.1128/jcm.02995-14
Lavine JS, Bjornstad ON, de Blasio BF, Storsaeter J (2012) Short-lived immunity against pertussis, age-specific routes of transmission, and the utility of a teenage booster vaccine. Vaccine 30(3):544–551. https://doi.org/10.1016/j.vaccine.2011.11.065
Stefanelli P (2019) Pertussis: identification, prevention and control. Adv Exp Med Biol 1183:127–36. https://doi.org/10.1007/5584_2019_408
European Centre for Disease Prevention and Control (2019) External quality assessment for the detection of Bordetella pertussis by PCR, 2018 – on behalf of EUPert-LabNet network. Stockholm, ECDC
Cherry JD (2005) The epidemiology of pertussis: a comparison of the epidemiology of the disease pertussis with the epidemiology of Bordetella pertussis infection. Pediatrics 115(5):1422–1427. https://doi.org/10.1542/peds.2004-2648
Zepp F, Heininger U, Mertsola J, Bernatowska E, Guiso N, Roord J et al (2011) Rationale for pertussis booster vaccination throughout life in Europe. Lancet Infect Dis 11(7):557–570. https://doi.org/10.1016/s1473-3099(11)70007-x
Esposito S, Stefanelli P, Fry NK, Fedele G, He Q, Paterson P et al (2019) Pertussis prevention: reasons for resurgence, and differences in the current acellular pertussis vaccines. Front Immunol 10:1344. https://doi.org/10.3389/fimmu.2019.01344
Wendelboe AM, Van Rie A, Salmaso S, Englund JA (2005) Duration of immunity against pertussis after natural infection or vaccination. Pediatr Infect Dis J 24(5 Suppl):S58-61. https://doi.org/10.1097/01.inf.0000160914.59160.41
Guiso N, Njamkepo E, Vié le Sage F, Zepp F, Meyer CU, Abitbol V et al (2007) Long-term humoral and cell-mediated immunity after acellular pertussis vaccination compares favourably with whole-cell vaccines 6 years after booster vaccination in the second year of life. Vaccine 25(8):1390–7. https://doi.org/10.1016/j.vaccine.2006.10.048
Mooi FR, Van Der Maas NA, De Melker HE (2014) Pertussis resurgence: waning immunity and pathogen adaptation - two sides of the same coin. Epidemiol Infect 142(4):685–694. https://doi.org/10.1017/s0950268813000071
Barkoff AM, Mertsola J, Pierard D, Dalby T, Hoegh SV, Guillot S et al (2019) Pertactin-deficient Bordetella pertussis isolates: evidence of increased circulation in Europe, 1998 to 2015. Euro Surveill: Bull Europeen sur les maladies transmissibles = Eur Commun Dis Bull 24(7) https://doi.org/10.2807/1560-7917.Es.2019.24.7.1700832.
Bart MJ, Harris SR, Advani A, Arakawa Y, Bottero D, Bouchez V et al (2014) Global population structure and evolution of Bordetella pertussis and their relationship with vaccination. mBio 5(2):e01074. https://doi.org/10.1128/mBio.01074-14
van Gent M, Heuvelman CJ, van der Heide HG, Hallander HO, Advani A, Guiso N et al (2015) Analysis of Bordetella pertussis clinical isolates circulating in European countries during the period 1998–2012. Eur J Clin Microbiol Infect Dis: Off Publ Eur Soc Clin Microbiol 34(4):821–830. https://doi.org/10.1007/s10096-014-2297-2
Zomer A, Otsuka N, Hiramatsu Y, Kamachi K, Nishimura N, Ozaki T et al (2018) Bordetella pertussis population dynamics and phylogeny in Japan after adoption of acellular pertussis vaccines. Microbial Genom 4(5); https://doi.org/10.1099/mgen.0.000180
Barkoff AM, Mertsola J, Pierard D, Dalby T, Hoegh SV, Guillot S et al (2018) Surveillance of circulating Bordetella pertussis strains in Europe during 1998 to 2015. J Clin Microbiol 56(5); https://doi.org/10.1128/jcm.01998-17
Weigand MR, Williams MM, Peng Y, Kania D, Pawloski LC, Tondella ML (2019) Genomic survey of Bordetella pertussis diversity, United States, 2000–2013. Emerg Infect Dis 25(4):780–783. https://doi.org/10.3201/eid2504.180812
Weigand MR, Pawloski LC, Peng Y, Ju H, Burroughs M, Cassiday PK et al (2018) Screening and genomic characterization of filamentous hemagglutinin-deficient Bordetella pertussis. Infect Immun 86(4); https://doi.org/10.1128/iai.00869-17
van Gent M, Bart MJ, van der Heide HG, Heuvelman KJ, Mooi FR (2012) Small mutations in Bordetella pertussis are associated with selective sweeps. PLoS ONE 7(9):e46407. https://doi.org/10.1371/journal.pone.0046407
Mooi FR, van Loo IH, van Gent M, He Q, Bart MJ, Heuvelman KJ et al (2009) Bordetella pertussis strains with increased toxin production associated with pertussis resurgence. Emerg Infect Dis 15(8):1206–1213. https://doi.org/10.3201/eid1508.081511
Xu Y, Liu B, Grondahl-Yli-Hannuksila K, Tan Y, Feng L, Kallonen T et al (2015) Whole-genome sequencing reveals the effect of vaccination on the evolution of Bordetella pertussis. Sci Rep 5:12888. https://doi.org/10.1038/srep12888
Octavia S, Maharjan RP, Sintchenko V, Stevenson G, Reeves PR, Gilbert GL et al (2011) Insight into evolution of Bordetella pertussis from comparative genomic analysis: evidence of vaccine-driven selection. Mol Biol Evol 28(1):707–715. https://doi.org/10.1093/molbev/msq245
Seppälä E, Kristoffersen AB, Bøås H, Vestrheim DF, Greve-Isdahl M, De Blasio BF et al (2021) Pertussis epidemiology and indirect impact of the childhood pertussis booster vaccinations, Norway, 1998–2019. Abstract, ESCAIDE
Zeddeman A, van Gent M, Heuvelman CJ, van der Heide HG, Bart MJ, Advani A et al (2014) Investigations into the emergence of pertactin-deficient Bordetella pertussis isolates in six European countries, 1996 to 2012. Euro Surveill: Bull Europeen sur les maladies transmissibles = Eur Commun Dis Bull 19(33) https://doi.org/10.2807/1560-7917.es2014.19.33.20881.
Khelef N, Danve B, Quentin-Millet MJ, Guiso N (1993) Bordetella pertussis and Bordetella parapertussis: two immunologically distinct species. Infect Immun 61(2):486–490. https://doi.org/10.1128/iai.61.2.486-490.1993
Diavatopoulos DA, Cummings CA, Schouls LM, Brinig MM, Relman DA, Mooi FR (2005) Bordetella pertussis, the causative agent of whooping cough, evolved from a distinct, human-associated lineage of B. bronchiseptica. PLoS Pathogens 1(4):e45. https://doi.org/10.1371/journal.ppat.0010045
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol 35(6):1547–1549. https://doi.org/10.1093/molbev/msy096
Yao K, Deng J, Ma X, Dai W, Chen Q, Zhou K et al (2020) The epidemic of erythromycin-resistant Bordetella pertussis with limited genome variation associated with pertussis resurgence in China. Expert Rev Vaccines 19(11):1093–9. https://doi.org/10.1080/14760584.2020.1831916
Letunic I, Bork P (2019) Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47(W1):W256–W259. https://doi.org/10.1093/nar/gkz239
Celentano LP, Massari M, Paramatti D, Salmaso S, Tozzi AE (2005) Resurgence of pertussis in Europe. Pediatr Infect Dis J 24(9):761–765. https://doi.org/10.1097/01.inf.0000177282.53500.77
Ntezayabo B, De Serres G, Duval B (2003) Pertussis resurgence in Canada largely caused by a cohort effect. Pediatr Infect Dis J 22(1):22–27. https://doi.org/10.1097/00006454-200301000-00009
Safarchi A, Octavia S, Wu SZ, Kaur S, Sintchenko V, Gilbert GL et al (2016) Genomic dissection of Australian Bordetella pertussis isolates from the 2008–2012 epidemic. J Infect 72(4):468–477. https://doi.org/10.1016/j.jinf.2016.01.005
Lefrancq N, Bouchez V, Fernandes N, Barkoff A-M, Bosch T, Dalby T et al (2021) Spatial dynamics and vaccine-induced fitness changes of Bordetella pertussis.
Bouchez V, Guillot S, Landier A, Armatys N, Matczak S, group tFpms et al (2021) Evolution of Bordetella pertussis over a 23-year period in France, 1996 to 2018. Eurosurveillance 26(37):2001213; https://doi.org/10.2807/1560-7917.ES.2021.26.37.2001213.
Clarke M, McIntyre PB, Blyth CC, Wood N, Octavia S, Sintchenko V et al (2016) The relationship between Bordetella pertussis genotype and clinical severity in Australian children with pertussis. J Infect 72(2):171–178. https://doi.org/10.1016/j.jinf.2015.11.004
Xu Z, Wang Z, Luan Y, Li Y, Liu X, Peng X et al (2019) Genomic epidemiology of erythromycin-resistant Bordetella pertussis in China. Emerg Microbes Infect 8(1):461–70. https://doi.org/10.1080/22221751.2019.1587315
Alai S, Ghattargi VC, Gautam M, Patel K, Pawar SP, Dhotre DP et al (2020) Comparative genomics of whole-cell pertussis vaccine strains from India. BMC Genomics 21(1):345. https://doi.org/10.1186/s12864-020-6724-8
Safarchi A, Saedi S, Octavia S, Sedaghatpour M, Bolourchi N, Tay CY et al (2021) Evolutionary genomics of recent clinical Bordetella pertussis isolates from Iran: wide circulation of multiple ptxP3 lineages and report of the first ptxP3 filamentous hemagglutinin-negative B. pertussis. Infect Genet Evol: J Mol Epidemiol Evol Genet Infect Dis 93:104970. https://doi.org/10.1016/j.meegid.2021.104970
Schmidtke A, Boney K, Martin S, Skoff T, Tondella ML, Tatti K (2012) Population diversity among Bordetella pertussis isolates, United States, 1935–2009. Emerg Infect Dis J 18(8):1248. https://doi.org/10.3201/eid1808.120082
Ring N, Abrahams JS, Bagby S, Preston A, MacArthur I (2019) How genomics is changing what we know about the evolution and genome of Bordetella pertussis. Adv Exp Med Biol 1183:1–17. https://doi.org/10.1007/5584_2019_401
Ma L, Caulfield A, Dewan K, Harvill E (2021) Pertactin-deficient Bordetella pertussis, vaccine-driven evolution, and reemergence of pertussis. Emerg Infect Dis J 27(6):1561. https://doi.org/10.3201/eid2706.203850
Safarchi A, Octavia S, Nikbin VS, Lotfi MN, Zahraei SM, Tay CY et al (2019) Genomic epidemiology of Iranian Bordetella pertussis: 50 years after the implementation of whole cell vaccine. Emerg Microbes Infect 8(1):1416–27. https://doi.org/10.1080/22221751.2019.1665479
Hiramatsu Y, Miyaji Y, Otsuka N, Arakawa Y, Shibayama K, Kamachi K (2017) Significant decrease in pertactin-deficient Bordetella pertussis isolates, Japan. Emerg Infect Dis J 23(4):699. https://doi.org/10.3201/eid2304.161575
Otsuka N, Han H-J, Toyoizumi-Ajisaka H, Nakamura Y, Arakawa Y, Shibayama K et al (2012) Prevalence and genetic characterization of pertactin-deficient Bordetella pertussis in Japan. PLoS ONE 7(2):e31985. https://doi.org/10.1371/journal.pone.0031985
Locht C, Carbonetti NH, Cherry JD, Damron FH, Edwards KM, Fernandez R et al (2020) Highlights of the 12th International Bordetella Symposium. Clin Infect Dis: Off Publ Infect Dis Soc Am 71(9):2521–2526. https://doi.org/10.1093/cid/ciaa651
Dorji D, Mooi F, Yantorno O, Deora R, Graham RM, Mukkur TK (2018) Bordetella Pertussis virulence factors in the continuing evolution of whooping cough vaccines for improved performance. Med Microbiol Immunol 207(1):3–26. https://doi.org/10.1007/s00430-017-0524-z
Bowden KE, Weigand MR, Peng Y, Cassiday PK, Sammons S, Knipe K et al (2016) Genome structural diversity among 31 Bordetella pertussis isolates from two recent U.S. whooping cough statewide epidemics. mSphere 1(3) https://doi.org/10.1128/mSphere.00036-16
Weigand MR, Peng Y, Loparev V, Batra D, Bowden KE, Burroughs M et al (2017) The history of Bordetella pertussis genome evolution includes structural rearrangement. J Bacteriol 199(8); https://doi.org/10.1128/jb.00806-16
Ring N, Abrahams JS, Jain M, Olsen H, Preston A, Bagby S (2018) Resolving the complex Bordetella pertussis genome using barcoded nanopore sequencing. Microbial Genomics 4(11):e000234. https://doi.org/10.1099/mgen.0.000234
Bouchez V, Brun D, Dore G, Njamkepo E, Guiso N (2011) Bordetella parapertussis isolates not expressing pertactin circulating in France. Clin Microbiol Infect: Off Publ Eur Soc Clin Microbiol Infect Dis 17(5):675–682. https://doi.org/10.1111/j.1469-0691.2010.03303.x
Barkoff AM, Mertsola J, Guillot S, Guiso N, Berbers G, He Q (2012) Appearance of Bordetella pertussis strains not expressing the vaccine antigen pertactin in Finland. Clin Vaccine Immunol 19(10):1703–1704. https://doi.org/10.1128/cvi.00367-12
Acknowledgements
We acknowledge the medical microbiological laboratories in Norway for submitting B. pertussis isolates or PCR-positive samples to the National Reference Laboratory at the Norwegian Institute of Public Health. We thank the Institut Pasteur teams for the curation and maintenance of BIGSdb-Pasteur databases at http://bigsdb.pasteur.fr/. LTB undertook this work as part of the European Public Health Microbiology Training Programme (EUPHEM), European Centre for Disease Prevention and Control (ECDC). An ECDC representative formally approved the manuscript before submission.
Funding
Open access funding provided by Norwegian Institute of Public Health (FHI) The study was funded fully by the Norwegian Institute of Public Health.
Author information
Authors and Affiliations
Contributions
DFV and LTB had the idea and formed the project, with input from MGI, AS, and OBB. RBR, TB, and MT performed the laboratory work, and OBB was responsible for the bioinformatics analysis. LTB and OBB interpreted all results. LTB wrote the first draft of the manuscript. All authors critically reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
This study was approved by the Regional Committees for Medical and Health Research Ethics in Norway (reference 82752).
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brandal, L.T., Vestrheim, D.F., Bruvik, T. et al. Evolution of Bordetella pertussis in the acellular vaccine era in Norway, 1996 to 2019. Eur J Clin Microbiol Infect Dis 41, 913–924 (2022). https://doi.org/10.1007/s10096-022-04453-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-022-04453-0