
Abstract
The aim of our study was to investigate phenotypic and genotypic features of streptococci misidentified (misID) as Streptococcus pneumoniae, obtained over 20 years from hospital patients in Poland. Sixty-three isolates demonstrating microbiological features typical for pneumococci (optochin susceptibility and/or bile solubility) were investigated by phenotypic tests, lytA and 16S rRNA gene polymorphism and whole-genome sequencing (WGS). All isolates had a 6-bp deletion in the lytA 3′ terminus, characteristic for Mitis streptococc and all but two isolates lacked the pneumococcal signature cytosine at nucleotide position 203 in the 16S rRNA genes. The counterparts of psaA and ply were present in 100% and 81.0% of isolates, respectively; the spn9802 and spn9828 loci were characteristic for 49.2% and 38.1% of isolates, respectively. Phylogenetic trees and networks, based on the multilocus sequence analysis (MLSA) scheme, ribosomal multilocus sequence typing (rMLST) scheme and core-genome analysis, clearly separated investigated isolates from S. pneumoniae and demonstrated the polyclonal character of misID streptococci, associated with the Streptococcus pseudopneumoniae and Streptococcus mitis groups. While the S. pseudopneumoniae clade was relatively well defined in all three analyses, only the core-genome analysis revealed the presence of another cluster comprising a fraction of misID streptococci and a strain proposed elsewhere as a representative of a novel species in the Mitis group. Our findings point to complex phylogenetic and taxonomic relationships among S. mitis-like bacteria and support the notion that this group may in fact consist of several distinct species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The group of Mitis streptococci [1, 2] encompasses species that significantly differ in their pathogenic potential. The best-known representative of the group, Streptococcus pneumoniae (pneumococcus), is responsible for a high burden of respiratory tract and invasive infections, especially in children and the elderly [3]. However, species such as Streptococcus pseudopneumoniae, Streptococcus mitis, Streptococcus oralis and others are typically non-pathogenic residents of the human nasopharynx, although invasive infections caused by these bacteria are also occasionally observed [2, 4]. The conventional microbiological identification of S. pneumoniae in clinical practice relies on two features: (1) susceptibility to optochin; (2) and solubility in sodium deoxycholate (bile), together with a specific morphology of colonies producing alpha-hemolysis on blood agar [5]. Reactivity with specific antibodies recognizing polysaccharide capsule (latex agglutination tests and the quellung reaction, i.e. capsular swelling) is another important feature of pneumococci; however, isolates of other closely related species occasionally also demonstrate these properties [6,7,8,9,10,11]. To complicate species identification issues, a small number of ‘true’ pneumococcal isolates present as optochin resistant and/or poorly soluble in bile [12,13,14]. Moreover, so-called rough pneumococci do not produce polysaccharide capsule, due to either mutations in the cps locus or the complete lack of this locus in certain lineages [15]. Misidentification may not only delay the proper treatment of a patient, but it also influences the correct estimation of the burden of disease caused by pneumococci and other viridians streptococci. In addition, as particular species in the Mitis group differ in the prevalence of resistance to antimicrobials, misidentification results in biased reporting of susceptibility levels in S. pneumoniae [11, 13, 16].
With the development of molecular techniques, DNA-based methods have been proposed to improve the identification of Mitis streptococci, mostly focusing on confirming or excluding identification of an isolate as S. pneumoniae. Initially, certain targets such as genes encoding pneumolysin (ply), autolysin (lytA), pneumococcal surface antigen A (psaA), conserved genes in the cps locus and spn9802 and spn9828 loci of unknown function were proposed as specific for S. pneumoniae [17,18,19,20,21]. However, their specificity was later questioned due to the presence of counterparts of some of these ‘pneumococcal’ genes in other Mitis streptococci [22, 23]. More specific approaches, which employed PCR-RFLP of lytA and the 16S rRNA genes [24, 25] and partial sequencing of sodA and rpoB [26, 27], proved to be more reliable for the purpose of identification. The correct selection of appropriate target gene(s) is important not only for the correct identification of isolates but also for culture-free detection of pneumococci in clinical materials, which often relies on PCR-based methods [28, 29]. Multilocus sequence typing (MLST), based on sequencing of seven loci encoding housekeeping genes of S. pneumoniae and identification of alleles and sequence types (STs) from allelic profiles with the web-accessible database (https://pubmlst.org/spneumoniae/), is considered to provide an unambiguous identification of an isolate as pneumococcus [8]. Multilocus sequence analysis (MLSA), based on the same principle but using different target loci, allows identification of species within the Mitis and other viridians streptococci [30]. These two approaches, however, are available only for specialized laboratories, both in terms of equipment and software as well as adequately trained personnel. The recent advances of whole-genome sequencing (WGS) technologies and increasing availability of WGS for laboratories have opened new possibilities also for identification purposes. In particular, ribosomal MLST (rMLST) that indexes variation of the 53 genes encoding bacterial ribosomal proteins has been proposed as a universal identification tool [31].
The National Reference Center for Bacterial Meningitis (NRCBM), Poland, has performed a systematic, country-wide, voluntary-based surveillance of invasive and respiratory tract infections caused by S. pneumoniae in Poland since 1997 (http://koroun.edu.pl/) and possesses an archival collection of pneumococcal isolates starting from the early 1990s. During its activity, the NRCBM occasionally received isolates identified as S. pneumoniae in clinical laboratories, which at the NRCBM were classified as other Mitis streptococci, usually negative in serotyping and/or presenting MLST profiles composed of new alleles, divergent from those characteristic for pneumococci. The aim of our study was to investigate these isolates using a genomic approach in order to better understand their mutual genetic relationships and position within the Mitis group, especially in the relation to S. pneumoniae.
Materials and methods
Bacterial isolates and patient data
Sixty-three misidentified streptococcal isolates (‘misID’ streptococci) from 22 centres in 20 cities were included in the study (Table 1). These isolates originated from the NRCBM collection including approximately 3100 respiratory tract and approximately 3600 invasive isolates received as S. pneumoniae from 1997 until the end of 2015 as well as from the archival collection of the laboratory of approximately 500 isolates from the early 1990s. Thus, misID streptococci accounted for approximately 1% of all pneumococcal isolates and approximately 2% of isolates from the respiratory tract, which constituted the principal source of misID isolates. In particular, among 63 misID streptococci, 29 were obtained from bronchoalveolar aspirates/lavages (BAL), 26 from sputum and six from throat swabs, and single isolates were derived from a central catheter and blood. Patients were aged from 13 to 92 years; information on age was not provided for five patients. Forty-one (65%) and 15 (24%) patients were male and female, respectively; in seven cases, sex was not reported. The male to female ratio was similar to the one observed for 5328 patients with S. pneumoniae infections in Poland in 2006–2015 (62% and 37%, respectively; p = 0.1).
Phenotypic tests
All misID isolates were retested with standard procedures used for S. pneumoniae identification, i.e. for bile solubility and optochin susceptibility (https://www.cdc.gov/meningitis/lab-manual/chpt08-id-characterization-streppneumo.pdf; 8th August 2019, date last accessed). Both these tests were performed at least twice. Antimicrobial susceptibility testing was performed as recommended by the European Committee on Antimicrobial Susceptibility Testing (EUCAST) using the viridans group streptococci breakpoints for penicillin, amoxicillin, ceftriaxone, clindamycin and vancomycin; and S. pneumoniae breakpoints for erythromycin, telithromycin, linezolid, chloramphenicol and rifampicin [32]; for ciprofloxacin, isolates with the MIC values > 4 mg/L were considered nonsusceptible as previously for S. pneumoniae [33].
DNA isolation and lytA and 16S rRNA genes-based identification
Total DNA was purified using the Genomic DNA Prep Plus kit (A&A Biotechnology, Gdynia, Poland) following the manufacturer’s instructions. The 3′ terminal part of the lytA gene was amplified as described [24] and analysed by sequencing of the amplified product. The PCR-RFLP with BsiHKAI restriction endonuclease (New England BioLabs, Hertfordshire, UK) was used to detect the A203C polymorphism in the 16S rRNA genes [25]. The R6 strain DNA was used as a positive control for PCR-RFLP.
Genomic sequencing and data analysis
Genomic sequencing was performed with MiSeq (Illumina, San Diego, CA) as an external service (GENOMED, Warsaw, Poland) with sequencing depth of at least 40×. Runs were assembled into contigs using the CLC software v9.0.1 (QIAGEN, Aarhus, Denmark). The presence of acquired antimicrobial resistance genes was verified using the ResFinder 3.1 database (https://cge.cbs.dtu.dk/services/ResFinder/; 8th August 2019, date last accessed). The online tool https://pubmlst.org/rmlst/ (8th August 2019, date last accessed) was used for the identification of rMLST-types (rSTs). Novel alleles of ribosomal protein gene loci and novel rMLST profiles found among studied isolates were submitted to the rMLST database. In silico MLST [34] was performed using the S. pneumoniae MLST database [35] (https://pubmlst.org/spneumoniae/; 8th August 2019, date last accessed).
To compile the reference set for the study, the following data were downloaded from GenBank (http://www.ncbi.nlm.nih.gov/genbank/, accessed the 17th December 2018): (1) complete genomes and whole-genome sequences of S. pseudopneumoniae and S. mitis; (2) whole-genome sequence of the 596553 strain, a potentially novel representative of the Mitis group [36]; (3) complete genomes of S. pneumoniae; (5) complete genomes of other representatives of Mitis group, including S. oralis, Streptococcus cristatus, Streptococcus peroris, Streptococcus australis, Streptococcus gordonii, Streptococcus infantis, Streptococcus parasanguinis and Streptococcus sanguinis. rSTs were verified using the rMLST database [35] (https://pubmlst.org/rmlst/; 8th August 2019, date last accessed), and strains with unique complete rMLST profiles were used in further analyses (26 S. pseudopneumoniae, 61 S. mitis, the 596553 strain, 40 S. pneumoniae and 8 other Mitis streptococci; Supplementary Table 1). The genomic sequences of studied isolates and reference strains were uploaded to a private instance of BIGSdb and used for MLSA, rMLST and gene presence/absence analyses with the BLAST tool using default parameters [37]. For MLSA, the scheme including seven loci as described elsewhere [30] was set up in BIGSdb. The rMLST-based phylogenetic analysis was performed using the MUSCLE algorithm [38] and the scheme available in the BIGSdb, excluding the BACT00062 locus (rpmG), which has paralogous genes in streptococcal genomes [31]; i.e., the scheme encompassed 52 loci. The concatenated alignments resulting from MLSA and rMLST analyses were used to construct neighbor-nets using the SplitsTree v.4 [39] and maximum likehood (ML) trees in MEGA-X [40] with 500 bootstrap replicas. Genome annotations were performed using Prokka [41], and the core genome was established using Roary [42]. Core-genome alignments were used to construct approximately-maximum-likelihood (AML) trees in FastTree [43, 44], which were visualized in FigTree (available at http://tree.bio.ed.ac.uk/software/figtree/; 18th February 2019, date last accessed).
The differences in distributions were evaluated with the chi-squared test, with the p value < 0.05 considered significant. The adjusted Wallace coefficient (AW) with confidence intervals (CI) as a measure of congruence between identification methods was calculated using the online tool Comparing Partitions at http://www.comparingpartitions.info/?link=Home (8th August 2019, date last accessed). An in silico DNA-DNA hybridization (dDDH) was carried out using the Genome-to-Genome Distance Calculator (GGDC) [45].
Accession numbers
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession VMKU00000000-VMNE00000000 in the BioProject PRJNA556140. The version described in this paper is version VMKU01000000-VMNE01000000.
Results
Phenotypic features of misID streptococci and antimicrobial resistance determinants
All 63 misID isolates presented at least one phenotypic feature typical for pneumococci, such as bile solubility (44 isolates) and optochin susceptibility (62 isolates, of which five repeatedly showed a borderline zone of inhibition of 14–15 mm) (Table 1). Antimicrobial susceptibility results are summarized in Table 2 for 62 isolates (a single isolate repeatedly did not grow under the test conditions, i.e. in cation-adjusted Mueller Hinton broth supplemented with 5% lysed horse blood). Thirty-one isolates (50%) demonstrated a multi-drug-resistant (MDR) phenotype; i.e., they were not susceptible to antimicrobials belonging to three or more classes tested. Analysis of genomic sequences (see below) using ResFinder revealed that 26 and nine isolates carried the erm(B) and mef(A) genes, respectively, including two isolates containing concomitantly both genes; two isolates had the cat gene and 32 isolates were positive for the tet(M) gene. Antimicrobial susceptibility phenotypes to erythromycin, clindamycin, chloramphenicol and tetracycline were generally in a good agreement with the distribution of acquired antimicrobial resistance genes (Table 1) with the exception of three erythromycin-susceptible isolates and a single tetracycline-susceptible isolate, harbouring presumably silent copies of the erm(B) and tet(M) genes. The analysis of the translated amino acid sequences of quinolone-determining regions (QRDR) in ParC revealed single isolates with S79R and S79I changes (Table 1); no mutations typically associated with quinolone nonsusceptibility were observed in the QRDR of GyrA.
Identification based on the polymorphism of the lytA and 16S rRNA genes
The 6-bp deletion in the terminal part of the lytA gene, specific for Mitis streptococci other than S. pneumoniae [24], was detected in all 63 isolates. Patterns observed in the PCR-RFLP of 16S rRNA genes were consistent with the presence of nucleotides other than the signature cytosine at the position 203, specific for S. pneumoniae [25] in all but two isolates, which presented mixed patterns. A subsequent detailed examination of sequencing genomic reads (see below) revealed the presence of both adenosine and cytosine at the position 203, suggesting the heterogeneity of 16S rRNA copies.
MLST, rMLST and distribution of the counterparts of psaA, ply and the spn9802 and spn9828 loci
Genomic sequencing and assembly yielded genomes ranging approximately from 1.85 to 2.28 Mb in size for investigated misID streptococcal isolates (Table 1). When in silico MLST was performed on obtained genomic sequences, alleles specific for S. pneumoniae were observed in 47 (74.6%) isolates, ranging from a single locus up to all seven loci (two isolates representing ST11884; Table 1). This ST was reported for the NTPn 138 isolate, which in our core-genome analysis clustered with S. pseudopneumoniae and not S. pneumoniae (see below). It is worth noting, however, that particular alleles in the profile of this ST (139-371-345-325-389-677-656) can be found in the association with several other STs of S. pneumoniae in the database. In the rMLST analysis, all 54 observed profiles were novel. Five rSTs were observed for more than a single isolate, including rSTs 106395 (5 isolates) and 23930, 106387, 106395 and 106415 (two isolates each). rMLST allele characteristics for misID streptococci were compared with the set of allelic profiles specific for 7270 rST of 27217 isolates of S. pneumoniae available at https://pubmlst.org/rmlst/ (as of the 12th May 2019). Alleles common for both misID streptococci and S. pneumoniae were found for all the rMLST loci (Supplementary Table 2). BLAST performed with sequences of genes proposed as specific for S. pneumoniae revealed the counterparts of psaA and ply in 100% and 81.0% of isolates, respectively, and the presence of the spn9802 and spn9828 loci in 49.2% and 38.1% of isolates, respectively (Table 1).
MLSA- and rMLST-based phylogenetic trees and networks
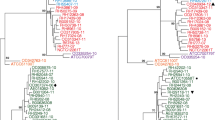
Concatenated and aligned genes from the MLSA and rMLST schemes were used to construct phylogenetic trees and networks that included misID streptococci and reference strains representing unique rSTs. In neighbor-nets based on both MLSA and rMLST, all S. pneumoniae strains formed a distinct cluster with relatively short branches. In particular, this group did not show clustering with any of the misID streptococci (Fig. 1a, b). This cluster was supported by 99–100% bootstrap values on the corresponding ML trees (Supplementary Fig. 1AB). The second cluster comprised 15 reference S. pseudopneumoniae strains and 28 misID streptococci on the MLSA-based neighbor-net and 15 reference S. pseudopneumoniae strains and 30 misID streptococci on the rMLST-based neighbor-net (Fig. 1a, b); the incongruence was observed for four misID streptococci and four reference strains (Fig. 1; Table 1 and Supplementary Table 1). The single misID isolate 12_5905 showed the closest clustering with the S. oralis Uo5 type strain [46]. However, considering the depth of these branches and their support, this isolate could not be unambiguously classified as S. oralis. The Uo5 strain and the 12_5905 isolate as well as S. cristatus, S. peroris, S. australis, S. gordonii, S. infantis, S. parasanguinis and S. sanguinis were clearly separated from other analysed strains and isolates, forming long branches in neighbor-nets and usually well-supported branches in ML trees. The remaining misID streptococci and S. mitis and S. pseudopneumoniae reference strains formed several branches, and the position of these was in several cases incongruent among MLSA and rMLST neighbor-nets. The 596553 strain proposed as a potentially novel species was present in this particular group. Generally, the AW coefficient between MLSA and rMLST for species identification of misID streptococci as S. pseudopneumoniae or S. mitis was 0.768 (CI 0.570–0.966).

Phylogenetic relationships among mitis group streptococci. a Neighbor-net, based on the MLSA sequences. b Neighbor-net, based on the rMLST sequences. Concatenated alignments were obtained in BIGSdb and analysed in SplitsTree. The S. pneumoniae and S. pseudopneumoniae clades marked by circles; reference strains of S. mitis (B6) and S. pseudopneumoniae (IS9374) with complete genomes available indicated by arrows; the 596553 strain marked by a triangle. The S. cristatus, S. peroris, S. australis, S. gordonii, S. infantis, S. parasanguinis and S. sanguinis branches trimmed to improve the readability of the figure. The M_, Ps_, SPN_ and NM_ prefixes included to mark reference strains of S. mitis, S. pseudopneumoniae, S. pneumoniae and the 596553 strain, respectively, from GenBank; the misID streptococci labelled with numbers according to Table 1. Reference strains and misID isolates with conflicting species identification by MLSA and rMLST marked by an asterisk and a hash, respectively
Core-genome analysis
Core-genome analysis was performed on misID streptococci (except for a single S. oralis-like isolate 12_5905); reference strains of S. pseudopneumoniae, S. mitis and S. pneumoniae; and the 596553 strain. After construction of an initial AML tree (data not shown), six strains belonging to major observed branches of S. pneumoniae were chosen. Finally, core-genome analysis on the group of 62 misID streptococci and 94 reference strains identified 523 common genes (Supplementary Table 3) and produced an alignment 561 519 bp in length, i.e. covering approximately 25–27% of genomes of the studied misID streptococci. The AML tree included the well-separated S. pneumoniae branch, the S. pseudopneumoniae cluster, comprising 31 misID streptococci and 16 reference strains, and several deep branches associated with S. mitis (Fig. 2). Ten reference strains, reported to the GenBank as S. pseudopneumoniae, were not included within this species (Supplementary Table 1), similarly to other reports [47]. The AW coefficient between the results of core-genome analysis with MLSA and rMLST for distinguishing S. pseudopneumoniae and S. mitis among the misID streptococci was 0.815 (CI 0.639–0.991) and 0.934 (CI 0.814–1.000), respectively. Among S. mitis, 25 misID streptococci formed a separate cluster that contained also the 596553 strain. Such clustering was not clearly apparent in earlier MLSA- and rMLST-based networks and trees (Fig. 1AB, Supplementary Fig. 1AB). The remaining six misID isolates were distributed among other S. mitis-like strains in the AML tree (Fig. 2). All isolates belonging to the S. pseudopneumoniae cluster harboured MLST alleles typical for S. pneumoniae while only 16 (51.6%) isolates of 596553-like and other S. mitis-like isolates showed the presence of such alleles (p = 0; Table 1). All misID representing S. pseudopneumoniae carried counterparts of ply and the 9808 locus while the presence of the 9828 locus was variable. In contrast, the presence of the ply counterparts was characteristic for 19 isolates (61.3%) of the 596553 cluster isolates and remaining S. mitis-like misID isolates, and all these lacked the 9808 and 9828 loci. Nonsusceptibility to penicillin was significantly more common among the 596553 cluster and other S. mitis-like isolates compared with S. pseudopneumoniae (p = 0.005). Such differences were not observed for other antimicrobial classes.

Phylogenomic relationships among misID streptococci, S. pneumoniae, S. mitis, S. pseudopneumoniae and the 596553 strain revealed by core-genome analysis. The approximately-maximum-likelihood tree was obtained in FastTree and visualized in FigTree. The S. pneumoniae, S. pseudopneumoniae and 96553 clades marked by rectangles; reference strains of S. mitis (B6) and of S. pseudopneumoniae (IS9374) with complete genomes available indicated by arrows; the 596553 strain marked by a triangle. The M_, Ps_, SPN_ and NM_ prefixes included to mark reference strains of S. mitis, S. pseudopneumoniae, S. pneumoniae and the 596553 strain, respectively, from GenBank; the misID streptococci labelled with numbers according to Table 1
Discussion
Correct species identification and understanding of the phylogenetic relationships within the Mitis group of streptococci still poses a challenge, despite several approaches addressing this issue over the years. This problem is associated with unusual mitis strains that are misidentified as S. pneumoniae (here proposed to be named misID streptococci). To our knowledge, this is the first attempt to characterize misID streptococci using genomic approaches at such scale. The analysed collection has certain features that make it especially interesting for the conducted analyses. Most isolates (56, i.e. 89%) were derived from clinically relevant materials, such as BAL, sputum and in a single-case blood, and they were collected over a relatively long time and localisation span. In this manner, risk of a potential bias due to repeated isolations was reduced, and high diversity of studied material was assured. All misID streptococci presented at least one and typically both phenotypic features used for differentiation of S. pneumoniae from other streptococci in microbiological laboratories, such as optochin susceptibility and bile solubility. While some strains of pneumococci are known to be optochin-resistant [12, 14], bile solubility is considered a principal characteristic of S. pneumoniae, and such observation in our study and other studies [48, 49] underlines the difficulty posed by such isolates for correct identification.
Nonsusceptibility to antimicrobials of main classes was very common in the analysed group and exceeded levels observed for S. pneumoniae in Poland ([50,49,50,53] and unpublished NRCBM data). Streptococci from the Mitis group are considered a reservoir and potential source of resistance genes for S. pneumoniae as indicated for the chromosomal pbp2b and pbp2x genes [54,53,56] and parC [57]. Also, the acquired resistance genes, such as erm(B), mef(A) and tet(M), are the same as major erythromycin and tetracycline resistance determinants in S. pneumoniae [58,57,60].
Several gene targets have been proposed as the basis for S. pneumoniae identification and detection in clinical material. In our collection, all misID streptococci harboured a 6-bp deletion in the lytA gene [24], indicating a very good performance of this test. However, both S. pseudopneumoniae with pneumococcal lytA and S. pneumoniae with lytA characteristic for Mitis streptococci have been observed [61]. Cytosine at nucleotide position 203 in the 16S rRNA genes is considered specific for the vast majority of pneumococci as it is replaced by adenosine in all other Mitis streptococci [25]. This test reliably distinguished misID streptococci in our study, with the exception of two isolates with mixed bases at this position. Several bacterial species carry more than one copy of the rRNA operon, and heterogeneity of copies of the 16S rRNA gene was observed earlier in S. oralis [62] and other species [63]. Among other proposed targets, psaA and ply were very common among the misID streptococci, and this observation was also made by others [22, 23, 64]. The spn9808 and spn9828 loci occurred ubiquitously among misID representing S. pseudopneumoniae but were absent among S. mitis-related isolates and thus could indeed exclude them as pneumococci.
Multilocus sequence-based approaches such as MLST, MLSA and rMLST were also evaluated as tools for distinguishing pneumococci and other Mitis streptococci. MLST following the S. pneumoniae scheme has been proposed as a method to reliably include or exclude isolates as pneumococcus [8]; however, in the current study, several isolates, especially among S. pseudopneumoniae, carried alleles’ characteristic for S. pneumoniae, up to a complete identification of two isolates as ST11884. Importantly, however, the single isolate NTP 138 representing this ST in the S. pneumoniae MLST database appears to be S. pneudopneumoniae in the core-genome analysis (Fig. 2) and as such should be removed from the database. In phylogenetic networks and trees, both MLSA and rMLST clearly separated the misID isolates from S. pneumoniae. The misID isolates showed the presence of several alleles from the rMLST scheme found also in pneumococci. This is different from the Neisseria genus, where some species shared some of rMLST alleles but not MLST alleles, and it was hypothesised that while the rMLST genes undergo recombination, metabolic genes from the MLST scheme evolve to specialize to particular niches [65]. The presence of a shared pool of both rMLST and MLST alleles in the misID streptococci and S. pneumoniae is consistent with a similarity of niches’ characteristic for both these groups. It may also indicate a relatively frequent horizontal transfer of genes between the misID streptococci and pneumococci [22] due to a natural competence common in the Mitis group [66].
The core-genome analysis, applied to investigate relatedness of misID streptococci to S. mitis, S. pseudopneumoniae, the 596553 strain and pneumococci, is considered the most reliable approach for such purposes for Mitis streptococci [67, 68]. The misID streptococci did not form a single cluster in the core-genome analysis but they were associated with S. pseudopneumoniae and with several branches of S. mitis. A similar grouping was also observed in a study on streptococcal isolates from respiratory tract and invasive infections, first considered atypical pneumococci [64]. In this study, with the use of MLSA, 61 S. pseudopneumoniae and 13 S. mitis were identified while 24 isolates could not be classified due to incomplete MLSA profiles. All these isolates, although collected in a single country (Spain), showed a remarkable diversity, similar to our observations. The core-genome analysis performed in our study revealed a clustering of the majority of misID streptococci associated with S. mitis into a separate group together with the 596553 strain. This strain was proposed to represent a novel species of streptococcus, based on a lack of clustering with the B6 strain of S. mitis, the IS7493 strain of S. pseudopneumoniae and the SPN032672 of S. pneumoniae genomes in single-nucleotide polymorphism (SNP) analysis and on 81% similarity to the most closely related species, S. pseudopneumoniae, revealed by protein-by-protein analysis [36]. The fact that we observed several epidemiologically independent isolates, clustering with the 596553 strain, further supports the idea that indeed this group might represent a novel species.
Two contrasting hypotheses were proposed concerning the evolution with the Mitis group. According to one, the common ancestor of Mitis streptococci was most similar to the current S. pneumoniae, and other representatives of the group adapted to a more commensal lifestyle by the loss of certain virulence-associated traits, resulting in a genome reduction [62]. Such reduction was not, however, apparent in our study. In contrast, misID genomes tended to be slightly larger (on average 2,152,263 ± 27,211 kb in comparison with 2,110,084 ± 29,809 kb observed for reference strains of S. pneumoniae used in this study; CI = 99%). The other hypothesis assumes that the S. pneumoniae species is relatively young and evolves due to its genome plasticity and acquisition of adaptive traits [68]; this hypothesis is in agreement with short branches and relative compactness of S. pneumoniae cluster in comparison with other Mitis streptococci. Such structure of the core-genome-based AML tree was observed here. It is important to note that misID streptococci did not show any clustering close to S. pneumoniae that might suggest their recent diversification from this species. However, features such as optochin susceptibility and bile solubility might have indeed been characteristic for a common ancestor of Mitis streptococci. While these properties have been lost by most of the members of this group, they have been preserved in a few lineages, in particular in S. pneumoniae and a few others, such as misID streptococci, which nowadays cause identification problems in clinical laboratories. It appears that the diversity of such organisms remains in a significant part unexplored, and more data are necessary to fully understand the relationships within this very particular group of bacteria.
References
Facklam R (2002) What happened to the streptococci: overview of taxonomic and nomenclature changes. Clin Microbiol Rev 15(4):613–630
Doern CD, Burnham CAD (2010) It’s not easy being green: the viridans group streptococci, with a focus on pediatric clinical manifestations. J Clin Microbiol 48(11):3829–3835. https://doi.org/10.1128/JCM.01563-10
World Health Organization (2019) Pneumococcal conjugate vaccines in infants and children under 5 years of age: WHO position paper. Wkly Epidemiol Rec 94:85–104r
Shelburne SA, Sahasrabhojane P, Saldana M, Yao H, Su X, Horstmann N, Thompson E, Flores AR (2014) Streptococcus mitis strains causing severe clinical disease in cancer patients. Emerg Infect Dis 20(5):762–771. https://doi.org/10.3201/eid2005.130953
Lund E, Henrichsen J (1978) Chapter XI. Laboratory diagnosis, serology and epidemiology of Streptococcus pneumoniae. Methods Microbiol 12:241–262
Martín-Galiano AJ, Balsalobre L, Fenoll A, de la Campa AG (2003) Molecular characterization of disease-associated streptococci of the mitis group that are optochin susceptible. Antimicrob Agents Chemother 47(10):3187–3194
Arbique JC, Poyart C, Trieu-Cuot P, Quesne G, Carvalho Mda G, Steigerwalt AG, Morey RE, Jackson D, Davidson RJ, Facklam RR (2004) Accuracy of phenotypic and genotypic testing for identification of Streptococcus pneumoniae and description of Streptococcus pseudopneumoniae sp. nov. J Clin Microbiol 42(10):4686–4696
Hanage WP, Kaijalainen T, Herva E, Saukkoriipi A, Syrjänen R, Spratt BG (2005) Using multilocus sequence data to define the pneumococcus. J Bacteriol 187(17):6223–6230
Balsalobre L, Hernández-Madrid A, Llull D, Martín-Galiano AJ, García E, Fenoll A, de la Campa AG (2006) Molecular characterization of disease-associated streptococci of the mitis group that are optochin susceptible. J Clin Microbiol 44(11):4163–4171
Leegaard TM, Bootsma HJ, Caugant DA, Eleveld MJ, Mannsåker T, Frøholm LO, Gaustad P, Høiby EA, Hermans PW (2010) Phenotypic and genomic characterization of pneumococcus-like streptococci isolated from HIV-seropositive patients. Microbiology 156(Pt 3):838–848. https://doi.org/10.1099/mic.0.035345-0
Simões AS, Sá-Leão R, Eleveld MJ, Tavares DA, Carriço JA, Bootsma HJ, Hermans PW (2010) Highly penicillin-resistant multidrug-resistant pneumococcus-like strains colonizing children in Oeiras, Portugal: genomic characteristics and implications for surveillance. J Clin Microbiol 48(1):238–246. https://doi.org/10.1128/JCM.01313-09
Phillips G, Barker R, Brogan O (1988) Optochin-resistant Streptococcus pneumoniae. Lancet 2(8605):281
Richter SS, Heilmann KP, Dohrn CL, Riahi F, Beekmann SE, Doern GV (2008) Accuracy of phenotypic methods for identification of Streptococcus pneumoniae isolates included in surveillance programs. J Clin Microbiol 46(7):2184–2188. https://doi.org/10.1128/JCM.00461-08
Pinto TC, Souza AR, de Pina SE, Costa NS, Borges Neto AA, Neves FP, Merquior VL, Dias CA, Peralta JM, Teixeira LM (2013) Phenotypic and molecular characterization of optochin-resistant Streptococcus pneumoniae isolates from Brazil, with description of five novel mutations in the atpC gene. J Clin Microbiol 51(10):3242–3249. https://doi.org/10.1128/JCM.01168-13
Hilty M, Wüthrich D, Salter SJ, Engel H, Campbell S, Sá-Leão R, de Lencastre H, Hermans P, Sadowy E, Turner P, Chewapreecha C, Diggle M, Pluschke G, McGee L, Eser ÖK, Low DE, Smith-Vaughan H, Endimiani A, Küffer M, Dupasquier M, Beaudoing E, Weber J, Bruggmann R, Hanage WP, Parkhill J, Hathaway LJ, Mühlemann K, Bentley SD (2014) Global phylogenomic analysis of nonencapsulated Streptococcus pneumoniae reveals a deep-branching classic lineage that is distinct from multiple sporadic lineages. Genome Biol Evol 6(12):3281–3294. https://doi.org/10.1093/gbe/evu263
Wester CW, Ariga D, Nathan C, Rice TW, Pulvirenti J, Patel R, Kocka F, Ortiz J, Weinstein RA (2002) Possible overestimation of penicillin resistant Streptococcus pneumoniae colonization rates due to misidentification of oropharyngeal streptococci. Diagn Microbiol Infect Dis 42(4):263–268
Corless CE, Guiver M, Borrow R, Edwards-Jones V, Fox AJ, Kaczmarski EB (2001) Simultaneous detection of Neisseria meningitidis, Haemophilus influenzae, and Streptococcus pneumoniae in suspected cases of meningitis and septicemia using real-time PCR. J Clin Microbiol 39(4):1553–1558
McAvin JC, Reilly PA, Roudabush RM, Barnes WJ, Salmen A, Jackson GW, Beninga KK, Astorga A, McCleskey FK, Huff WB, Niemeyer D, Lohman KL (2001) Sensitive and specific method for rapid identification of Streptococcus pneumoniae using real-time fluorescence PCR. J Clin Microbiol 39(10):3446–3451
Morrison KE, Lake D, Crook J, Carlone GM, Ades E, Facklam R, Sampson JS (2000) Confirmation of psaA in all 90 serotypes of Streptococcus pneumoniae by PCR and potential of this assay for identification and diagnosis. J Clin Microbiol 38(1):434–437
Park HK, Lee SJ, Yoon JW, Shin JW, Shin HS, Kook JK, Myung SC, Kim W (2010) Identification of the cpsA gene as a specific marker for the discrimination of Streptococcus pneumoniae from viridans group streptococci. J Med Microbiol 59(Pt 10):1146–1152. https://doi.org/10.1099/jmm.0.017798-0
Suzuki N, Seki M, Nakano Y, Kiyoura Y, Maeno M, Yamashita Y (2005) Discrimination of Streptococcus pneumoniae from viridans group streptococci by genomic subtractive hybridization. J Clin Microbiol 43(9):4528–4534
Whatmore AM, Efstratiou A, Pickerill AP, Broughton K, Woodard G, Sturgeon D, George R, Dowson CG (2000) Genetic relationships between clinical isolates of Streptococcus pneumoniae, Streptococcus oralis, and Streptococcus mitis: characterization of “atypical” pneumococci and organisms allied to S. mitis harboring S. pneumoniae virulence factor-encoding genes. Infect Immun 68(3):1374–1382
Neeleman C, Klaassen CH, Klomberg DM, de Valk HA, Mouton JW (2004) Pneumolysin is a key factor in misidentification of macrolide-resistant Streptococcus pneumoniae and is a putative virulence factor of S. mitis and other streptococci. J Clin Microbiol 42(9):4355–4357
Llull D, López R, García E (2006) Characteristic signatures of the lytA gene provide a basis for rapid and reliable diagnosis of Streptococcus pneumoniae infections. J Clin Microbiol 44(4):1250–1256
Scholz CF, Poulsen K, Kilian M (2012) Novel molecular method for identification of Streptococcus pneumoniae applicable to clinical microbiology and 16S rRNA sequence-based microbiome studies. J Clin Microbiol 50(6):1968–1973. https://doi.org/10.1128/JCM.00365-12
Kawamura Y, Whiley RA, Shu SE, Ezaki T, Hardie JM (1999) Genetic approaches to the identification of the mitis group within the genus Streptococcus. Microbiology 145(Pt 9):2605–2613
Glazunova OO, Raoult D, Roux V (2010) Partial recN gene sequencing: a new tool for identification and phylogeny within the genus Streptococcus. Int J Syst Evol Microbiol 60(Pt 9):2140–2148. https://doi.org/10.1099/ijs.0.018176-0
Carvalho Mda G, Tondella ML, McCaustland K, Weidlich L, McGee L, Mayer LW, Steigerwalt A, Whaley M, Facklam RR, Fields B, Carlone G, Ades EW, Dagan R, Sampson JS (2007) Evaluation and improvement of real-time PCR assays targeting lytA, ply, and psaA genes for detection of pneumococcal DNA. J Clin Microbiol 45(8):2460–2466
Book M, Lehmann LE, Zhang X, Stüber F (2013) Monitoring infection: from blood culture to polymerase chain reaction (PCR). Best Pract Res Clin Anaesthesiol 27(2):279–288. https://doi.org/10.1016/j.bpa.2013.06.010
Bishop CJ, Aanensen DM, Jordan GE, Kilian M, Hanage WP, Spratt BG (2009) Assigning strains to bacterial species via the internet. BMC Biol 7:3. https://doi.org/10.1186/1741-7007-7-3
Jolley KA, Bliss CM, Bennett JS, Bratcher HB, Brehony C, Colles FM, Wimalarathna H, Harrison OB, Sheppard SK, Cody AJ, Maiden MC (2012) Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology 158(Pt 4):1005–1015. https://doi.org/10.1099/mic.0.055459-0
The European Committee on Antimicrobial Susceptibility Testing (2018) Breakpoint tables for interpretation of MICs and zone diameters Version 8
Sadowy E, Izdebski R, Skoczyńska A, Gniadkowski M, Hryniewicz W (2005) High genetic diversity of ciprofloxacin-nonsusceptible isolates of Streptococcus pneumoniae in Poland. Antimicrob Agents Chemother 49(5):2126–2129
Enright MC, Spratt BG (1998) A multilocus sequence typing scheme for Streptococcus pneumoniae: identification of clones associated with serious invasive disease. Microbiology 144(Pt 11):3049–3060
Jolley KA, Bray JE, Maiden MCJ (2018) Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res 3:124. https://doi.org/10.12688/wellcomeopenres.14826.1
Kirkeleite IØ, Bohlin J, Scheffer L, Weme ET, Vestrheim DF (2018) Draft genome sequence of a potentially novel Streptococcus species belonging to the Streptococcus mitis group. Genome Announc 6(26). https://doi.org/10.1128/genomeA.00620-18
Jolley KA, Maiden MC (2010) BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797. https://doi.org/10.1093/nar/gkh340
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23(2):254–267
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35(6):1547–1549
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30(14):2068–2069. https://doi.org/10.1093/bioinformatics/btu153
Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, Fookes M, Falush D, Keane JA, Parkhill J (2015) Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31(22):3691–3693. https://doi.org/10.1093/bioinformatics/btv421
Price MN, Dehal PS, Arkin AP (2009) FastTree: computing large minimum-evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26(7):1641–1650. https://doi.org/10.1093/molbev/msp077
Price MN, Deha PS, Arkin AP (2010) FastTree 2 - approximately maximum-likelihood trees for large alignments. PLoS One 5(3):e9490. https://doi.org/10.1371/journal.pone.0009490
Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M (2013) Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. https://doi.org/10.1186/1471-2105-14-60
Reichmann P, Nuhn M, Denapaite D, Brückner R, Henrich B, Maurer P, Rieger M, Klages S, Reinhard R, Hakenbeck R (2011) Genome of Streptococcus oralis strain Uo5. J Bacteriol 193(11):2888–2889. https://doi.org/10.1128/JB.00321-11
Jensen A, Scholz CF, Kilian M (2016) Re-evaluation of the taxonomy of the Mitis group of the genus Streptococcus based on whole genome phylogenetic analyses, and proposed reclassification of Streptococcus dentisani as Streptococcus oralis subsp. dentisani comb. nov., Streptococcus tigurinus as Streptococcus oralis subsp. tigurinus comb. nov., and Streptococcus oligofermentans as a later synonym of Streptococcus cristatus. Int J Syst Evol Microbiol 66(11):4803–4820. https://doi.org/10.1099/ijsem.0.001433
Ikryannikova LN, Lapin KN, Malakhova MV, Filimonova AV, Ilina EN, Dubovickaya VA, Sidorenko SV, Govorun VM (2011) Misidentification of alpha-hemolytic streptococci by routine tests in clinical practice. Infect Genet Evol 11(7):1709–1715. https://doi.org/10.1016/j.meegid.2011.07.010
Ing J, Mason EO, Kaplan SL, Lamberth LB, Revell PA, Luna RA, Hulten KG (2012) Characterization of nontypeable and atypical Streptococcus pneumoniae pediatric isolates from 1994 to 2010. J Clin Microbiol 50(4):1326–1330. https://doi.org/10.1128/JCM.05182-11
Skoczyńska A, Hryniewicz W (2003) Genetic relatedness, antibiotic susceptibility, and serotype distribution of Streptococcus pneumoniae responsible for meningitis in Poland, 1997-2001. Microb Drug Resist 9(2):175–182
Skoczyńska A, Kadłubowski M, Waśko I, Fiett J, Hryniewicz W (2007) Resistance patterns of selected respiratory tract pathogens in Poland. Clin Microbiol Infect 13(4):377–383
Skoczyńska A, Sadowy E, Bojarska K, Strzelecki J, Kuch A, Gołębiewska A, Waśko I, Foryś M, van der Linden M, Hryniewicz W, Participants of laboratory-based surveillance of community acquired invasive bacterial infections (BINet) (2011) The current status of invasive pneumococcal disease in Poland. Vaccine 29(11):2199–2205. https://doi.org/10.1016/j.vaccine.2010.09.100
Skoczyńska A, Kuch A, Sadowy E, Waśko I, Markowska M, Ronkiewicz P, Matynia B, Bojarska A, Wasiak K, Gołębiewska A, van der Linden M, Hryniewicz W, Participants of a laboratory-based surveillance of community acquired invasive bacterial infections (BINet) (2015) Recent trends in epidemiology of invasive pneumococcal disease in Poland. Eur J Clin Microbiol Infect Dis 34(4):779–787. https://doi.org/10.1007/s10096-014-2283-8
Dowson CG, Hutchison A, Brannigan JA, George RC, Hansman D, Liñares J, Tomasz A, Smith JM, Spratt BG (1989) Horizontal transfer of penicillin-binding protein genes in penicillin-resistant clinical isolates of Streptococcus pneumoniae. Proc Natl Acad Sci U S A 86(22):8842–2846
Laible G, Spratt BG, Hakenbeck R (1991) Interspecies recombinational events during the evolution of altered PBP2× genes in penicillin-resistant clinical isolates of Streptococcus pneumoniae. Mol Microbiol 5(8):1993–2002
van der Linden M, Otten J, Bergmann C, Latorre C, Liñares J, Hakenbeck R (2017) Insight into the diversity of penicillin-binding protein 2x alleles and mutations in Viridans streptococci. Antimicrob Agents Chemother 61(5):e02646–e02616. https://doi.org/10.1128/AAC.02646-16
Balsalobre L, Ferrándiz MJ, Liñares J, Tubau F, de la Campa AG (2003) Viridans group streptococci are donors in horizontal transfer of topoisomerase IV genes to Streptococcus pneumoniae. Antimicrob Agents Chemother 47(7):2072–2081
Chopra I, Roberts M (2001) Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol Mol Biol Rev 65(2):232–260. https://doi.org/10.1128/MMBR.65.2.232-260.2001
Izdebski R, Sadowy E, Fiett J, Grzesiowski P, Gniadkowski M, Hryniewicz W (2007) Clonal diversity and resistance mechanisms in tetracycline-nonsusceptible Streptococcus pneumoniae isolates in Poland. Antimicrob Agents Chemother 51(4):1155–1163
Fyfe C, Grossman TH, Kerstein K, Sutcliffe J (2016) Resistance to macrolide antibiotics in public health pathogens. Cold Spring Harb Perspect Med 6(10). https://doi.org/10.1101/cshperspect.a025395
Simões AS, Tavares DA, Rolo D, Ardanuy C, Goossens H, Henriques-Normark B, Linares J, de Lencastre H, Sá-Leão R (2016) lytA-based identification methods can misidentify Streptococcus pneumoniae. Diagn Microbiol Infect Dis 85(2):141–148. https://doi.org/10.1016/j.diagmicrobio.2016.03.018
Kilian M, Poulsen K, Blomqvist T, Håvarstein LS, Bek-Thomsen M, Tettelin H, Sørensen UB (2008) Evolution of Streptococcus pneumoniae and its close commensal relatives. PLoS One 3:e2683
Acinas SG, Marcelino LA, Klepac-Ceraj V, Polz MF (2004) Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J Bacteriol 186(9):2629–2635
Rolo D, Simões AS, Domenech A, Fenoll A, Liñares J, de Lencastre H, Ardanuy C, Sá-Leão R (2013) Disease isolates of Streptococcus pseudopneumoniae and non-typeable S. pneumoniae presumptively identified as atypical S. pneumoniae in Spain. PLoS One 8(2):e57047. https://doi.org/10.1371/journal.pone.0057047
Bennett JS, Jolley KA, Earle SG, Corton C, Bentley SD, Parkhill J, Maiden MC (2012) A genomic approach to bacterial taxonomy: an examination and proposed reclassification of species within the genus Neisseria. Microbiology 158(Pt 6):1570–1580. https://doi.org/10.1099/mic.0.056077-0
Salvadori G, Junges R, Morrison DA, Petersen FC (2019) Competence in Streptococcus pneumoniae and close commensal relatives: mechanisms and implications. Front Cell Infect Microbiol 9:94. https://doi.org/10.3389/fcimb.2019.00094
Rasmussen LH, Dargis R, Højholt K, Christensen JJ, Skovgaard O, Justesen US, Rosenvinge FS, Moser C, Lukjancenko O, Rasmussen S, Nielsen XC (2016) Whole genome sequencing as a tool for phylogenetic analysis of clinical strains of Mitis group streptococci. Eur J Clin Microbiol Infect Dis 35(10):1615–1625. https://doi.org/10.1007/s10096-016-2700-2
Velsko IM, Perez MS, Richards VP (2019) Resolving phylogenetic relationships for Streptococcus mitis and Streptococcus oralis through core- and pan-genome analyses. Genome Biol Evol 11(4):1077–1087. https://doi.org/10.1093/gbe/evz049
Acknowledgements
We thank Polish microbiologists for providing us isolates used in this study and Radosław Izdebski for his kind advice with PROKKA and ROARY. This publication made use of the PubMLST website (https://pubmlst.org/) sited at the University of Oxford. The development of that website was funded by the Wellcome Trust.
Funding
This work was supported by the grant 2013/10/M/NZ2/00259 from Narodowe Centrum Nauki (NCN), Poland.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Isolates were obtained as a part of routine activity of the NRCBM and were analysed anonymously in a retrospective manner. Ethical approval and informed consent were thus not required.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sadowy, E., Bojarska, A., Kuch, A. et al. Relationships among streptococci from the mitis group, misidentified as Streptococcus pneumoniae. Eur J Clin Microbiol Infect Dis 39, 1865–1878 (2020). https://doi.org/10.1007/s10096-020-03916-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-020-03916-6