
Abstract
The bronchial microbiome in severe COPD during stability and exacerbation in patients chronically colonised by Pseudomonas aeruginosa (PA), has not been defined. Our objective was to determine the characteristics of the bronchial microbiome of severe COPD patients colonised and not colonised by P. aeruginosa and its changes during exacerbation. COPD patients with severe disease and frequent exacerbations were categorised according to chronic colonisation by P. aeruginosa. Sputum samples were obtained in stability and exacerbation, cultured, and analysed by 16S rRNA gene amplification and pyrosequencing. Sixteen patients were included, 5 of them showing chronic colonisation by P. aeruginosa. Pseudomonas genus had significantly higher relative abundance in stable colonised patients (p = 0.019), but no significant differences in biodiversity parameters were found between the two groups (Shannon, 3 (2–4) vs 3 (2–3), p = 0.699; Chao1, 124 (77–159) vs 140 (115–163), p = 0.364). In PA-colonised patients bronchial microbiome changed to a microbiome similar to non-PA-colonised patients during exacerbations. An increase in the relative abundance over 20 % during exacerbation was found for Streptococcus, Pseudomonas, Moraxella, Haemophilus, Neisseria, Achromobacter and Corynebacterium genera, which include recognised potentially pathogenic microorganisms, in 13 patients colonised and not colonised by P. aeruginosa with paired samples. These increases were not identified by culture in 5 out of 13 participants (38.5 %). Stable COPD patients with severe disease and PA-colonised showed a similar biodiversity to non-PA-colonised patients, with a higher relative abundance of Pseudomonas genus in bronchial secretions. Exacerbation in severe COPD patients showed the same microbial pattern, independently of previous colonisation by P. aeruginosa.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sputum has traditionally been used for the identification of bronchial infection by potentially pathogenic microorganisms (PPMs) in exacerbated chronic obstructive pulmonary disease (COPD) patients, and positive cultures are found in a significant proportion of patients in this clinical setting, Haemophilus influenzae, Streptococcus pneumoniae and Moraxella catarrhalis being the bacteria most often recovered [1, 2]. P. aeruginosa is a cause of exacerbation in severe COPD patients [3–8], and studies that have collected sequential samples of bronchial secretions from patients with advanced disease have demonstrated that the acquisition of this PPM is in most cases associated with the appearance of exacerbation symptoms [9]. Advanced respiratory disease is considered a significant risk factor for P. aeruginosa infection when exacerbation symptoms appear, together with previous hospitalisations and antibiotics or steroid courses, and the use of antibiotic therapy targeting this PPM is recommended in current guidelines when these determinants are identified [10].
Persistence of positive cultures of bronchial secretions for P. aeruginosa after an exacerbation occurs in a minority of severe COPD patients, and in most cases is a transient phenomenon [11]. However, long-term persistence of P. aeruginosa as a chronic colonising strain has been reported in up to one fifth of COPD patients with a sputum culture positive for this PPM [9, 11, 12], and more commonly in patients who had associated bronchiectasis [13]. COPD patients with severe disease who have had previous positive cultures for P. aeruginosa have a worse prognosis when exacerbation symptoms appear [8], and the use of antibiotic therapy active against P. aeruginosa in these patients is common in current practice [12], but open to debate [14].
Currently available culture-based techniques are not suitable for the identification of up to 80 % of microorganisms inhabiting mucosal surfaces [15, 16]. Bacteria that are part of the respiratory microbiome are difficult to culture by conventional methods, and false-negative results are often found in the face of obligate anaerobes and when bacterial loads are low [17]. The application of pyrosequencing to PCR-amplified 16S rRNA gene has taken the study of microbial diversity to an unprecedented level of detail [18–20]. Culture-independent microbiological techniques have demonstrated that the bronchial tree is not sterile during health and have documented significant changes in the respiratory microbiome in chronic lung diseases [21]. In this study, we aimed to determine the differences in the bronchial microbiota in severe COPD patients with chronic colonisation by P. aeruginosa and in patients non-colonised by this PPM and the change of this microbiota when an exacerbation appears, identifying the microorganisms that overgrow during the episode. Bronchial secretions were examined by amplification and pyrosequencing of the 16S rRNA gene, and patients with and without chronic colonisation by this PPM were compared, both in stability periods and during exacerbation.
Materials and methods
Design and population
We enrolled a cohort of stable severe COPD outpatients who had been diagnosed according to the criteria of the Global Initiative for Chronic Obstructive Lung Disease (GOLD) [22] and who had reported more than two exacerbations per year, between January and December 2010. Patients were examined at baseline in a stable condition from >8 weeks, and their sociodemographic data, smoking habits, respiratory symptoms, treatments, sputum microbiological characteristics and lung function were recorded. Participants were followed for 6 months after this baseline assessment, and follow-up visits were scheduled every 3 months, or when exacerbation symptoms appeared. Every visit included the recovery of spontaneous sputum samples that were processed for microbiological cultures and culture-independent microbiological techniques. Detailed exclusion criteria are provided in the online supplementary file. Acute episodes of increased breathlessness, sputum production and/or purulence appearing during follow-up and treatment with antibiotics and/or oral corticosteroids by a physician were considered exacerbations [23]. Patients with positive cultures for P. aeruginosa in three or more consecutive sputum cultures obtained during a period of 6 months before enrolment in the study were considered to be chronically colonised by this PPM [24, 25]. The present study was reviewed and approved by the local research ethics committee in Catalonia, Spain, and written informed consent was obtained from all subjects.
Clinical variables, sputum collection and microbiology
Sociodemographic data were recorded at recruitment. Patients answered an epidemiological questionnaire that covered smoking habits, respiratory symptoms, previous exacerbations and treatments. Spontaneous sputum was collected from every patient under clinical stability and during exacerbation, before the administration of antibiotic therapy. Details on the clinical variables and microbiological processing are provided in the supplementary file.
Samples and DNA extraction
Sputum samples diluted with dithiothreitol were incubated at 37 °C for 15 min and centrifuged for 10 min at 4 °C. The pellet was resuspended in 1 ml of a lysis buffer containing 100 U/ml mutanolysin, 47,700 U/ml lysozime and 2 U/ml lysostaphin, and was incubated for 30 min at 37 °C. DNA was extracted using a QIAamp DNA Blood Midi kit (Qiagen, Helden, Germany) according to the manufacturer’s instructions and eluted in 200 μl of sterile water. DNA was quantified in the Nanodrop ND-1000 Spectrophotometer (NanoDrop Techologies, Wilmington, DE, USA) and stored at –80 °C.
PCR amplification of the V1–V3 region of the 16S rRNA gene
The hypervariable regions V1, V2 and V3 of the 16S rRNA gene were amplified with E8 forward (AGAGTTTGATCMTGGCTCAG) and B530 reverse (CCGCGGCKGCTGGCAC) primers and with 12 different barcodes. The PCRs were carried out in a volume of 40 μl with Biomix (Bioline, London, UK) and 10 mM of each primer, and were performed on the Mastercycler® pro thermal cycler (Eppendorf, Hamburg, Germany). After amplification, the products were visualised in 2 % agarose gels. Amplified product was purified using NucleoFast® 96 PCR Clean-Up kit (Macherey-Nagel, Düren, Germany), eluted in 28 μl of PCR-grade water and quantified with Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, Life Technologies, Carlsbad, CA, USA). Twelve samples with different barcode sequences were pooled in equimolar amounts into a single tube and pyrosequencing was carried out using Roche 454 GS-FLX System Titanium Chemistry.
Library analysis sequences and microbiome accession numbers
16S rRNA raw sequences were analysed using the mothur software package 1.27 [26] to remove sequences that were less than 200 bp or greater than 520 bp in length and chimeras. The Quantitative Insights into Microbial Ecology (QIIME) pipeline [27] was used for sequence processing to obtain taxonomic information. A taxonomic classification was performed and operational taxonomic units (OTUs) present in the samples at 97 % identity were determined.
Bacterial 16S rDNA data sets from this study are accessible in the European Nucleotide Archive under the study accession number PRJEB4144, available at http://www.ebi.ac.uk/ena/data/view/PRJEB4144, with the sample accession numbers ERS255709–739.
Statistical analysis
Statistical analyses were performed using SPSS statistical software package version 18 (SPSS, Chicago, IL, USA), R package (http://www.r-project.org), using the VEGAN library to cluster and construct abundance heat maps and LEfSe to identify differences between groups [28]. Results for categorical variables are expressed as absolute and relative frequencies and results for continuous variables as means and standard deviations (SD), or as medians and percentiles 25–75 (P25–P75) when the distribution was not normal.
Bacterial biodiversity was assessed through the Chao1 estimator [29] and Shannon [30] index, which estimate the richness and homogeneity of the microbiome respectively. Both indices were calculated after subsampling with QIIME to avoid sequencing effort bias. The beta-diversity Bray–Curtis dissimilarity index [31] was used to make principal correspondence analysis.
Variability of the bacterial flora between consecutive samples of the same patient was expressed as a percentage of change of relative abundance (relative abundance consecutive sample – relative abundance baseline sample × 100). Baseline variability for all genera was calculated by the comparison of consecutive samples obtained during stability from the same patient, to establish the expected variability range under stable conditions for severe COPD patients. Variability of the bacterial flora when exacerbation appears was equally expressed as percentage of change of relative abundance, using the previous baseline sample as the reference (relative abundance exacerbation sample – relative abundance baseline sample × 100). Statistical tests used in the study were two-sided, and a p value of 0.05 or less was reported to be statistically significant.
Results
Patients’ characteristics
Sixteen severe COPD patients, 6 of them at GOLD stage III (37.5 %) and 10 at stage IV (62.5 %), were included in the study. All of them were former smokers with an average age of 71 (SD 6) years (Table 1). Five patients had criteria of chronic P. aeruginosa colonisation, and participants were classified into two groups according to colonisation by this PPM. Microbiological data of both groups were compared and the variability in exacerbation was assessed for PA-colonised and non-PA-colonised patients, using the previous available results in stability as the reference.
Bronchial microbiome
Microbiological cultures were performed in sputum samples obtained during stability (n = 14) and exacerbation periods (n = 15). Cultures were positive for PPMs in 12 stability (86 %) and 10 exacerbation samples (67 %), P. aeruginosa and H. influenzae being the most frequent isolated PPMs in the studied patients (Table 2). Culture-independent analyses were also performed in all sputum samples obtained during stability and exacerbation and the average number of sequences per sample obtained after filtering was 4,695. Most prevalent phyla in bronchial microbiome were Proteobacteria (50 %) and Firmicutes (31 %), followed by Actinobacteria (8 %), Bacteroidetes (6 %) and Fusobacteria (3 %). One hundred twenty-eight different OTUs were obtained at genus level (supplementary file: Figure 1), and after removing the OTUs present in only one sample, the remaining ones were used for further analysis.
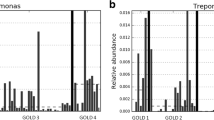
The relative abundance of specific genera in samples from PA-colonised and non-PA-colonised patients were compared, during both stability and exacerbation periods, to assess the effect of this colonisation on the remaining bronchial flora. Statistically significant differences were only found in the Pseudomonas genus, which had a higher relative abundance in PA-colonised patients in both the stable and the exacerbated state (Fig. 1, p = 0.019 and p = 0.003 respectively, Mann–Whitney U test). The biodiversity of bacterial communities in sputum samples obtained during stability measured with Chao1 richness estimator and Shannon index value showed no statistically significant differences between PA-colonised and non-PA-colonised groups (Shannon index, 3 (2–4) vs 3 (2–3), p = 0.669; Chao index, 124 (77–159) vs 140 (115–163), p = 0.364; Mann–Whitney U test). Similar results were obtained for the samples obtained during an exacerbation (Shannon index, 4 (2–4) vs 4 (2.5–5), p = 0.595; Chao index, 165 (93–196) vs 163 (70–193), p = 0.679, Mann–Whitney U test).

Relative abundance of Pseudomonas genus a under stability and b during exacerbation in PA-colonised and non-PA-colonised patients. Solid line represents the mean and dashed line the median
Rarefaction curves were calculated for PA-colonised and non-PA-colonised patients using the observed species metric (supplementary file: Figure 2) and showed that the number of sequenced taxa was enough to estimate total population diversity in both groups. Differences between microbial communities in both groups of patients were assessed by principal coordinates analyses with the Bray–Curtis beta-diversity metric, with samples obtained during stable (Fig. 2a) and exacerbation periods (Fig. 2b). Genera with the highest relative abundance were plotted in this analysis, and two clusters were observed in samples obtained during a stability period, corresponding to PA-colonised or non-PA-colonised samples, with Pseudomonas, Corynebacterium and Moraxella as the most abundant genera in colonised patients. During exacerbation, samples from PA-colonised and from non-PA-colonised patients did not cluster separately. The Bray–Curtis metric was also used after pooling all patients for clustering together patients colonised by P. aeruginosa during stability (PS) and exacerbation (PE); and stable (S) and exacerbated (E) non-PA-colonised COPD patients, according to genera composition (Fig. 3). In this analysis the bacterial community obtained during an exacerbation in PA-colonised patients showed a closer similarity to the microbiome of non-PA-colonised severe COPD participants, in both stable and exacerbated states, than to the microbiome recovered from PA-colonised patients during their stability periods. When most abundant genera in the four clinical situations were represented in a heat map, a range of genera showed low relative abundance but high prevalence, which included Veillonella, Actinomyces, Granulicatella, Neisseria, Prevotella, Tannerella, Gemella, Rothia and Achromobacter, with Streptococcus as the most abundant genus in all clinical situations (Fig. 4). Haemophilus, Moraxella and Pseudomonas genera, which include the main PPM in COPD, were also frequently recovered, but showed a high variability among the four assessed clinical situations. Pseudomonas genus was the most common in stable PA-colonised patients, Haemophilus being the genus more often found in non-PA-colonised patients and during exacerbation, independently of the previous history of P. aeruginosa colonisation.

Principal coordinate analysis with Bray–Curtis dissimilarity index. a Samples from stability and b samples of exacerbation. Red dots represent colonised patients, and blue dots non-PA-colonised patients

Cluster dendrogram with Bray–Curtis dissimilarity index. Samples were combined depending on their clinical situation and clustered with the Bray–Curtis index, which takes values between 0 and 1 (0 meaning that samples share all the genera and 1 meaning samples do not share any). PS colonised by P. aeruginosa under stability; PE colonised by P. aeruginosa during exacerbation; S non-colonised by P. aeruginosa under stability; E non-colonised by P. aeruginosa during exacerbation)

Heat map showing the most abundant genera in the four groups of samples. Columns represent the groups and rows the genera whose relative abundance is >1 % in at least one sample. The relative abundance of each genus is represented by the colour key
Variability in bronchial microbiome during exacerbation
Baseline variability of the bronchial microbiome in severe COPD was determined calculating the percentage of variation for all examined genera in two sputum samples obtained consecutively (>2 weeks) during stability from 2 patients (relative abundance consecutive sample – relative abundance baseline sample × 100), and was below 20 % for all the genera studied (n = 96; Fig. 5a). Changes over this value were accordingly considered abnormal for subsequent analysis. The same approach was followed for the calculation of the variability of every genus in the bronchial flora when exacerbation appears in 13 patients with paired samples from stability and exacerbation. Genera with increases over 20 % in exacerbations included Streptococcus, Pseudomonas, Moraxella, Haemophilus, Neisseria, Achromobacter and Corynebacterium, all of which include recognised PPMs. The increase observed for specific genera when exacerbation symptoms appear paralleled a decrease of the same magnitude in the relative abundance for genera unrelated to this exacerbation, without major changes in the remaining bronchial microbiota. Exacerbation-related variability in PA-colonised and non-PA-colonised patients was similar, and did not show statistically significant differences in the genera that showed increases in their relative abundance over 20 % (p >0.05, Chi-squared test; Fig. 5b, c).

Percentage of change in the relative abundance of genera. Only genera with a percentage of change over 20 % in at least one patient are represented. a Percentage of variability in the genera observed in consecutive samples recovered in stable situation (n = 2); b Percentage of variability in exacerbation in patients colonised by P. aeruginosa using the baseline stability sample as the reference (n = 5); c Percentage of variability in exacerbation in non-PA-colonised patients (n = 7). Dark blue: Neisseria; orange: Achromobacter; purple: Streptococcus; light blue: Pseudomonas; green: Moraxella; pink: Haemophilus; grey: Corynebacterium
Increases over 20 % in specific genera identified by 16SrRNA gene pyrosequencing were compared with culture results during exacerbation in the same sample, in order to determine the sensitivity of microbiological cultures for detecting the identified changes in the bronchial microbiome. In 5 out of 13 patients (38.5 %) microbial cultures did not show PPMs whose genera increased over 20 % from baseline at pyrosequencing. Three of these patients had negative sputum cultures during exacerbation and 1 patient showed a positive culture for P. aeruginosa, while an increase over 20 % was detected for Moraxella genus at pyrosequencing without significant changes in Pseudomonas genus abundance. In the fifth patient, an increase in Achromobacter genus was shown by the culture-independent technique, while the sputum culture was positive for Stenothropomonas maltophilia and Alcaligenes spp. In 2 exacerbated patients positive sputum cultures showed a PPM previously recovered during stability. These patients were considered to be colonised, and their pyrosequencing results did not show an increase in abundance over 20 % for any genera (Table 3).
Discussion
This study analysed the microbiome of severe COPD patients through amplification and pyrosequencing of the 16S rRNA gene. Assessing sputum samples obtained during stability from patients colonised by P. aeruginosa and non-PA-colonised patients, we observed that the microbiome of PA-colonised COPD patients showed an expected increase in the Pseudomonas genus. Differences in the bronchial microbiome between PA-colonised and non-PA-colonised patients disappeared during exacerbation, a clinical situation that prompted a change in the microbiome of patients chronically colonised by this PPM to a bacterial flora equivalent to that found in exacerbated non-PA-colonised patients. Besides, we found that some genera increased their relative abundance during exacerbation, including Streptococcus, Pseudomonas, Moraxella, Haemophilus, Neisseria, Achromobacter and Corynebacterium, all of which include recognised PPMs, and this change was equivalent in PA-colonised and non-PA-colonised patients. Simultaneous sputum cultures did not identify genera increasing their abundance in one third of the exacerbations appearing in severe COPD patients.
We found that the bronchial tree of severe COPD patients shows a wide range of prevalent genera, in agreement with previous reports in healthy subjects and patients with moderate disease [18, 19, 32]. Thus, the bacterial diversity in the lower airway in severe COPD individuals without signs of infection was much higher than previously anticipated, with a complex bacterial community that includes Streptococcus, Haemophilus, Moraxella, Veillonella, Actinomyces, Granulicatella, Neisseria, Prevotella, Tannerella, Gemella, Rothia and Achromobacter as the main recovered genera. Pseudomonas was the most common genus in stable patients who have shown previous colonisation by P. aeruginosa, and Haemophilus is the genus more often found in non-PA-colonised severe patients.
In spite of the chronic presence of P. aeruginosa in the bronchial tree of a subgroup of severe COPD patients, we found no statistically significant differences in Chao1 and Shannon biodiversity parameters between PA-colonised and non-PA-colonised patients, suggesting that the presence of P. aeruginosa in the respiratory tract of these patients does not modify the diversity of the bronchial microbiome. The conservation of a diverse microbiota is related to epithelial integrity, immunoregulation and colonisation resistance [19, 33–35] and shifts in the microbial community composition may compromise respiratory health and contribute to disease progression. Recent studies based on culture-independent sequencing technologies have reported well-defined microbiome changes in the mucosa of diverse chronic diseases, such as cystic fibrosis and inflammatory bowel diseases, among others [36]. The lack of differences in the bronchial microbiome observed in PA-colonised and non-PA-colonised patients in our study suggests that the bronchial colonisation by this PPM is not associated with the appearance of significant alterations in the bronchial flora in severe COPD patients.
We found that the bacterial community obtained during exacerbation in patients colonised by P. aeruginosa changes to a flora showing a close similarity with the microbiome found in non-PA-colonised severe COPD patients during exacerbation, suggesting that acute episodes in PA-colonised patients might be attributable to common PPMs instead of the colonising PPM. In severe COPD, P. aeruginosa may be recovered from sputum cultures obtained during stability periods [3–8]. Patients with persistent colonisation by P. aeruginosa have a worse prognosis than non-colonised patients when exacerbation symptoms appear [8], and require prompt treatment. Accordingly, antibiotic treatments targeting P. aeruginosa are currently recommended for severe patients with frequent episodes of exacerbation with this clinical profile [10]. In adults with COPD this approach is justified by the features that this disease shares with cystic fibrosis, in which P. aeruginosa causes chronic colonisation and is associated with significant morbidity and mortality [37–39]. Extrapolating observations from cystic fibrosis to COPD must be done with caution, however, because the carriage patterns of P. aeruginosa differ substantially in the two diseases [11, 40, 41]. Our findings suggest that exacerbation symptoms in PA-colonised patients are mainly related to PPMs that are a frequent cause of exacerbation in non-PA-colonised COPD patients rather than P. aeruginosa infection.
The variability in the abundance of genera in bronchial secretions observed during stability and exacerbation periods was assessed in our study, and stable severe COPD patients showed variabilities below 20 % for all genera in the absence of exacerbation symptoms. When exacerbation appears, this value increases well above the stability reference level for genera such as Haemophilus, Moraxella and Streptococcus, all of which include pathogenic species. These findings confirm that most cases of infectious exacerbation in severe COPD patients are due to PPMs, which are a common cause of acute symptoms in COPD, without significant differences between PA-colonised and non-PA-colonised patients. This finding suggests that antibiotic treatments prescribed for exacerbations in PA-colonised patients need to target common PPMs instead of the colonising PPM in the first instance. Interestingly, the increases in relative abundance observed in specific genera during exacerbation periods in the present study paralleled clear decreases of a similar magnitude in other genera that also included PPMs, without significant changes in the baseline flora found in the bronchial tree, again with a similar pattern in PA-colonised and non-PA-colonised patients.
The exacerbation-related changes identified in the bronchial microbiome during exacerbation were not identified by simultaneous microbiological cultures in one third of the patients in our study. Although traditional microbiological culture is the gold standard technique for the identification of bronchial colonisation [42], it has some limitations. A significant number of microorganisms do not grow in selective cultures, and fast-growing bacteria can mask other clinically important bacteria in the sample [43]. PCR amplification of 16S rRNA gene identifies the real composition of the lung microbiome [42] and can help us to understand the association between the microbiome changes and the acute stages of the disease.
Limitations of the study are the sample size, the use of sputum samples for the assessment of bronchial secretions and the absence of testing for viral infection. Enrolled patients were representative of the hospital-managed population with severe COPD, and did not suffer from severe comorbidities. Sputum was used to examine the bronchial microbiome because these samples can be obtained easily and non-invasively and the guidelines for the procedure include quality criteria. Although the sputum may be contaminated by bacterial flora of the oropharynx and the oral cavity, the microbiome composition that we have found in severe COPD patients is quite similar to that of previous reports in healthy controls and COPD patients based on the use of bronchial brushing and bronchoalveolar lavage for the sampling of bronchial secretions [18, 19]. We assumed, accordingly, that sputum is a representative sample for the study of the microbiome of bronchial secretions in severe COPD patients, with the additional advantage that the results obtained can be easily correlated with sputum culture. Viral infection in COPD patients can modify the bronchial microbiome [44] and changes in the microbiome detected during exacerbation may be partly influenced by the coexistence of infective virus in the bronchial tree.
Conclusions
Our data confirm that a rich bronchial microbiome is found in bronchial secretions from stable COPD patients with severe disease, which includes genera with well-known PPMs that are unusual in the normal population. In severe patients colonised by P. aeruginosa the bacterial community showed differences with regard to non-colonised patients in their stability periods, but at times of exacerbation their microbial profile changed to a flora that included PPMs and was equivalent to the bronchial microbiome found in non-colonised patients during exacerbation. These findings argue against the need for differentiated antibiotic approaches to exacerbation in severe COPD patients colonised by P. aeruginosa, a suggestion that must be confirmed in larger cohorts of patients.
References
Hill AT, Campbell EJ, Hill SL, Bayley DL, Stockley RA (2000) Association between airway bacterial load and markers of airway inflammation in patients with stable chronic bronchitis. Am J Med 109(4):288–295
Sethi S, Evans N, Grant BJ, Murphy TF (2002) New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Engl J Med 347(7):465–471
Soler N, Torres A, Ewig S, Gonzalez J, Celis R, El-Ebiary M, Hernandez C, Rodriguez-Roisin R (1998) Bronchial microbial patterns in severe exacerbations of chronic obstructive pulmonary disease (COPD) requiring mechanical ventilation. Am J Respir Crit Care Med 157(5 Pt 1):1498–1505
Eller J, Ede A, Schaberg T, Niederman MS, Mauch H, Lode H (1998) Infective exacerbations of chronic bronchitis: relation between bacteriologic etiology and lung function. Chest 113(6):1542–1548
Rosell A, Monso E, Soler N, Torres F, Angrill J, Riise G, Zalacain R, Morera J, Torres A (2005) Microbiologic determinants of exacerbation in chronic obstructive pulmonary disease. Arch Intern Med 165(8):891–897
Lieberman D (2003) Pseudomonal infections in patients with COPD: epidemiology and management. Am J Respir Med 2(6):459–468
Garcia-Vidal C, Almagro P, Romani V, Rodriguez-Carballeira M, Cuchi E, Canales L, Blasco D, Heredia JL, Garau J (2009) Pseudomonas aeruginosa in patients hospitalised for COPD exacerbation: a prospective study. Eur Respir J 34(5):1072–1078
Miravitlles M, Espinosa C, Fernandez-Laso E, Martos JA, Maldonado JA, Gallego M (1999) Relationship between bacterial flora in sputum and functional impairment in patients with acute exacerbations of COPD. Study Group of Bacterial Infection in COPD. Chest 116(1):40–46
Rakhimova E, Wiehlmann L, Brauer AL, Sethi S, Murphy TF, Tummler B (2009) Pseudomonas aeruginosa population biology in chronic obstructive pulmonary disease. J Infect Dis 200(12):1928–1935
Woodhead M, Blasi F, Ewig S, Huchon G, Ieven M, Ortqvist A, Schaberg T, Torres A, van der Heijden G, Verheij TJ (2005) Guidelines for the management of adult lower respiratory tract infections. Eur Respir J 26(6):1138–1180
Murphy TF, Brauer AL, Eschberger K, Lobbins P, Grove L, Cai X, Sethi S (2008) Pseudomonas aeruginosa in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 177(8):853–860
Martinez-Solano L, Macia MD, Fajardo A, Oliver A, Martinez JL (2008) Chronic Pseudomonas aeruginosa infection in chronic obstructive pulmonary disease. Clin Infect Dis 47(12):1526–1533
Pomares X, Monton C, Espasa M, Casabon J, Monso E, Gallego M (2011) Long-term azithromycin therapy in patients with severe COPD and repeated exacerbations. Int J Chron Obstruct Pulmon Dis 6:449–456
Murphy TF (2008) The many faces of Pseudomonas aeruginosa in chronic obstructive pulmonary disease. Clin Infect Dis 47(12):1534–1536
Hayashi H, Sakamoto M, Benno Y (2002) Phylogenetic analysis of the human gut microbiota using 16S rDNA clone libraries and strictly anaerobic culture-based methods. Microbiol Immunol 46(8):535–548
Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD, Dore J (1999) Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol 65(11):4799–4807
Sibley CD, Grinwis ME, Field TR, Eshaghurshan CS, Faria MM, Dowd SE, Parkins MD, Rabin HR, Surette MG (2011) Culture enriched molecular profiling of the cystic fibrosis airway microbiome. PLoS One 6(7):e22702
Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, Bushman FD, Collman RG (2011) Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med 184(8):957–963
Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, Young VB, Toews GB, Curtis JL, Sundaram B, Martinez FJ, Huffnagle GB (2011) Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One 6(2):e16384
Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, Davies J, Ervine A, Poulter L, Pachter L, Moffatt MF, Cookson WO (2010) Disordered microbial communities in asthmatic airways. PLoS One 5(1):e8578
Armougom F, Bittar F, Stremler N, Rolain JM, Robert C, Dubus JC, Sarles J, Raoult D, La Scola B (2009) Microbial diversity in the sputum of a cystic fibrosis patient studied with 16S rDNA pyrosequencing. Eur J Clin Microbiol Infect Dis 28(9):1151–1154
Updated 2013 Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. www.goldcopd.com
Anthonisen NR, Manfreda J, Warren CP, Hershfield ES, Harding GK, Nelson NA (1987) Antibiotic therapy in exacerbations of chronic obstructive pulmonary disease. Ann Intern Med 106(2):196–204
Canton R, Cobos N, de Gracia J, Baquero F, Honorato J, Gartner S, Alvarez A, Salcedo A, Oliver A, Garcia-Quetglas E (2005) Antimicrobial therapy for pulmonary pathogenic colonisation and infection by Pseudomonas aeruginosa in cystic fibrosis patients. Clin Microbiol Infect 11(9):690–703
Vendrell M, de Gracia J, Olveira C, Martinez MA, Giron R, Maiz L, Canton R, Coll R, Escribano A, Sole A (2008) Diagnosis and treatment of bronchiectasis. Spanish Society of Pneumology and Thoracic Surgery. Arch Bronconeumol 44(11):629–640
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75(23):7537–7541
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7(5):335–336
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12(6):R60
Chao A (1984) Nonparametric estimation of the number of classes in a population. Scand J Stat 11:265–270
Shannon CE (1997) The mathematical theory of communication. 1963. MD Comput 14(4):306–317
Bray JR, Curtis JT (1957) An ordination of the upland forest communities of southern Wisconsin. Ecol Monogr 27:325–349
Morris A, Beck JM, Schloss PD, Campbell TB, Crothers K, Curtis JL, Flores SC, Fontenot AP, Ghedin E, Huang L, Jablonski K, Kleerup E, Lynch SV, Sodergren E, Twigg H, Young VB, Bassis CM, Venkataraman A, Schmidt TM, Weinstock GM (2013) Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med 187(10):1067–1075
Curtis JL, Freeman CM, Hogg JC (2007) The immunopathogenesis of chronic obstructive pulmonary disease: insights from recent research. Proc Am Thorac Soc 4(7):512–521
Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, Roca J, Dore J (2006) Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 55(2):205–211
Sartor RB (2008) Therapeutic correction of bacterial dysbiosis discovered by molecular techniques. Proc Natl Acad Sci U S A 105(43):16413–16414
Frank DN, Zhu W, Sartor RB, Li E (2011) Investigating the biological and clinical significance of human dysbioses. Trends Microbiol 19(9):427–434
Burns JL, Gibson RL, McNamara S, Yim D, Emerson J, Rosenfeld M, Hiatt P, McCoy K, Castile R, Smith AL, Ramsey BW (2001) Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J Infect Dis 183(3):444–452
Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL (2002) Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol 34(2):91–100
West SE, Zeng L, Lee BL, Kosorok MR, Laxova A, Rock MJ, Splaingard MJ, Farrell PM (2002) Respiratory infections with Pseudomonas aeruginosa in children with cystic fibrosis: early detection by serology and assessment of risk factors. JAMA 287(22):2958–2967
Dal Negro R, Micheletto C, Tognella S, Visconti M, Turati C (2008) Tobramycin nebulizer solution in severe COPD patients colonized with Pseudomonas aeruginosa: effects on bronchial inflammation. Adv Ther 25(10):1019–1030
Orriols R, Roig J, Ferrer J, Sampol G, Rosell A, Ferrer A, Vallano A (1999) Inhaled antibiotic therapy in non-cystic fibrosis patients with bronchiectasis and chronic bronchial infection by Pseudomonas aeruginosa. Respir Med 93(7):476–480
Han MK, Huang YJ, Lipuma JJ, Boushey HA, Boucher RC, Cookson WO, Curtis JL, Erb-Downward J, Lynch SV, Sethi S, Toews GB, Young VB, Wolfgang MC, Huffnagle GB, Martinez FJ (2012) Significance of the microbiome in obstructive lung disease. Thorax 67(5):456–463
Morgan XC, Huttenhower C (2012) Chapter 12: Human microbiome analysis. PLoS Comput Biol 8(12):e1002808
Molyneaux PL, Mallia P, Cox MJ, Footitt J, Willis-Owen SA, Homola D, Trujillo-Torralbo MB, Elkin S, Kon OM, Cookson WO, Moffatt MF, Johnston SL (2013) Outgrowth of the bacterial airway microbiome after rhinovirus exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 188(10):1224–1231
Acknowledgements
We thank Michael Maudsley for providing an outline for this manuscript and support in editing and journal styling.
This work was supported by Sociedad Catalana de Neumología; Fundació Catalana de Neumología; Fundació Parc Tauli; Marató de TV3; Sociedad Española de Neumología y Cirugía Torácica; Ministerio de Economía y Competitividad (Spain; SAF2012-31187); Generalitat Valenciana (Spain; Prometeo/2009/092) and Centro de Investigación Biomédica en Red de Enfermedades Respiratorias (CIBERES). CIBERES is an initiative of the Instituto de Salud Carlos III. VPB has a postdoctoral contract in health research improvement “Sara Borrell” from the Instituto de Salud Carlos III (ISCIII).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Andrés Moya and Eduard Monsó contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 118 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Millares, L., Ferrari, R., Gallego, M. et al. Bronchial microbiome of severe COPD patients colonised by Pseudomonas aeruginosa . Eur J Clin Microbiol Infect Dis 33, 1101–1111 (2014). https://doi.org/10.1007/s10096-013-2044-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-013-2044-0