
Abstract
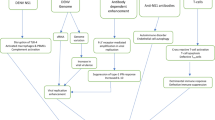
Toxoplasma gondii has a highly clonal genetic structure classified into three major genetic types, I, II, and III, plus additional recombinant and atypical strains. In humans, type I and atypical strains usually associate with severe toxoplasmosis. Type II strains, predominantly identified in European countries and the United States, correlate with a differential course of toxoplasmosis. During pregnancy, the important protective role of the placenta against maternal–fetal T. gondii transmission has been reported. T. gondii preferentially colonizes extravillous trophoblasts as compared to syncytiotrophoblasts. The latter compartment was suggested to act as the real barrier to the fetal dissemination of T. gondii. Alterations in immune response to particular T. gondii strains were observed. Higher transcription levels of IP-10, IL-1β, IL-6, IL-10, IL-12 cytokines, and NF-κB translocation to the nucleus were more often documented for type II strains than type I strains. Since the induction of IL-12 during type II infection was Myd88-dependent, the involvement of Toll-like receptors (TLRs) in the immunity against these strains was suggested. Differential expression of TLRs depends on placental cell types and gestational age. The expression of TLR2 and TLR4 in the first trimester of pregnancy was reported only for villous cytotrophoblasts and extravillous trophoblasts, but not for syncytiotrophoblasts. The involvement of single-nucleotide polymorphisms (SNPs) in the TLR genes in infectious pathogenicity, including toxoplasmic retinochoroiditis, points at a possible involvement of TLR alterations in immunity against T. gondii. We conclude that studies on TLR contributions in the maternal–fetal transmission of particular parasite strains and congenital toxoplasmosis are warranted.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Toxoplasma gondii genotypes contribute differentially to the course of toxoplasmosis
Toxoplasma gondii has a highly clonal genetic structure, with three major genetic types, I, II, and III, plus additional recombinant and atypical strains [1–4]. The three archetypic lineages were predominantly observed in North America and Europe, whereas more divergent genotypes were identified in French Guiana, Mexico, and Brazil [2, 5, 6].
Combined clinical and in vitro studies showed that outbreaks of toxoplasmosis presented with asymptomatic to symptomatic courses related to the genotypes involved. Differential virulence of T. gondii strains was observed for mice infected with parasites of particular lineages [7, 8]. Genotype I of T. gondii is most virulent for mice, inducing extensive parasite dissemination and sudden murine death. In contrast, genotype II causes non-fatal infection with much less tachyzoite dissemination [7]. However, strain-specific virulence varied between hosts, as was reported for mice and rats [8, 9]. Hence, it was suggested that T. gondii strains virulent for mice might not lead to a similar course of toxoplasmosis in humans.
Studies of humans with T. gondii mainly included cases of congenital toxoplasmosis and often originated in immune-deficient patients [7, 10–12]. Less frequent reports show symptomatic acquired toxoplasmosis in immune-competent patients [2, 5, 7]. T. gondii type II strains, identified predominantly in the populations of some European countries and the United States, were reported to generate congenital toxoplasmosis, including lethal infection, severe neuro-ocular involvement, isolated chorioretinitis, and/or latent toxoplasmosis [7]. However, type II strains were also isolated from benign or latent cases of toxoplasmosis, whereas type I and atypical strains are usually from severe cases [7, 13].
An atypical multi-locus T. gondii genotype identified from the amniotic fluid of a 29-year-old pregnant woman of Caucasian origin who was native to France was reported to cause severe congenital toxoplasmosis with bilateral ventricular enlargement and calcifications [1]. For at least 5 of 11 patients with laboratory-diagnosed toxoplasmosis living in Patam, a village near the French Guianan border, mouse inoculation or polymerase chain reaction (PCR) showed one atypical strain of T. gondii causing differential disease courses [5]. Eight immune-competent adults showed multi-visceral toxoplasmosis, leading to one death. One neonate and one fetus had lethal congenital toxoplasmosis and one child had symptomatic toxoplasmosis [5]. Hence, factors other than perceived strain virulence were suggested to influence the clinical presentation of toxoplasmosis in people living in an Amazonian rain forest [5].
The genotyping of 88 T. gondii isolates from immune-compromised patients in France showed a lack of significant differences in the distribution of parasite strains between patients diversified accordingly to cause of immunosuppression, site of infection, and outcome [14]. Hence, among immune-compromised patients, host factors were suggested as being co-correlated with toxoplasmosis development [14]. However, another study reported atypical T. gondii strains to have given rise to an outbreak of congenital toxoplasmosis and being responsible for more severe clinical courses of ocular toxoplasmosis in Brazilian children as compared to Europeans [15].
Toxoplasma gondii genotypes differentially transmit through the placenta
Despite the fact that differential immune responses resulted from infection with various T. gondii strains, only a few studies reported that the parasite maternal–fetal transmission through the placenta might depend on strain variations. However, the protective role against congenital infections with T. gondii was reported for the placenta and its particular compartments.
In a BALB/c mice model, animals were inoculated 30 days before breeding with T. gondii ME49 (type II) or M7741 (type III) strains. The same mice were re-infected 12 and 15 days after pregnancy with other strains (M7741 or ME49, respectively) and did not develop congenital infections [16]. Mice which were primary infected during pregnancy had no fetal infection as well, although their placentas were T. gondii-positive. Hence, the placenta played an important protective role against the maternal–fetal transmission of T. gondii [16]. No fetal infection developed despite the fact that parasites could be detected in placentas, kidneys, spleens, livers, and hearts of the BALB/c mice [17]. A higher frequency of infected placentas was observed at later stages of pregnancy, which has been correlated with the phagocytic efficiency of the placental tissues, possibly related to the stage of pregnancy as well [18, 19].
Studies with first-trimester human placental explants described the role of two anatomical placental interfaces [the villous trophoblast (fetal cells) and the extravillous trophoblast (EVT, maternal cells)] in the maternal–fetal transmission of T. gondii [20]. Similar to other pathogens, T. gondii preferentially colonized the EVT and not the relatively resistant syncytiotrophoblasts (ST). The multi-parasite vacuoles were primarily identified within subsyncytial cytotrophoblasts, possibly representing transsyncytial T. gondii transmission. As only single parasites were observed in ST, this placental compartment was suggested to act as a barrier to the fetal dissemination of T. gondii [20]. The comparison between three T. gondii strains showed no significant differences in their capacity to infect placental interfaces; only a slightly slower replication rate of the type II strain was observed [20]. Another study reported that BeWo choriocarcinoma cells went into apoptosis after infection with type II rather than type I strains. In this case, host cell death versus parasite death might reflect differential strain abilities to infect the placenta with varying degrees of virulence [21].
An important protective function was reported for IFN-γ, the key regulator of anti-T. gondii immunity [19]. Pregnant transgenic IFN-γ knock-out (GKO) B6 (susceptible) and BALB/c (resistant) mice infected with T. gondii showed higher parasite numbers in the uterus and placenta than wild-type (WT) mice [19]. In addition, also, murine genetic susceptibility to parasite infection was shown to be a key factor. For instance, fetal infection was observed only in GKO B6 and not among GKO BALB/c, WT B6, and WT BALB/c mice [19].
Toxoplasma gondii genotypes induce differential immune responses
In addition to the apparently differential virulence of various T. gondii genotypes, studies also showed altered immune responses against particular genotypes (Table 1) [22–25]. T. gondii-infected WT murine microglial cells showed variable kinetics of pro-inflammatory cytokine expression dependent on the parasite strain [22]. Higher and sustainable responses including elevated expression levels of IP-10 and IL-12b were observed in case of T. gondii II and III strains, but not for type I strains. The observed fluctuations in cytokine expression were time-dependent, and strain-dependent alterations of anti-apoptotic genes were minimal [22]. Oppositely, the infection of human neuroepithelioma cells with T. gondii type I strains caused bigger changes in the expression levels of a higher number of inflammation- and apoptosis-related genes than that observed for types II and III [25]. Macrophage immune responses to infection with T. gondii types I and II strains also showed a 2- to 3-fold elevated IL-12 gene expression [26]. Additionally, the induction of IL-12 expression after infection with type I strains did not involve Myd88 signaling, whereas this was clearly Myd88-dependent with type II strains.
The ME49 strain activated ERK1/2 and p38 MAPK in an Myd88-dependent manner and enhanced the expression of IL-12, which the RH strain failed to do [26]. Various signaling pathways involved in the immunity to T. gondii types I and II strains might affect the differential virulence of parasites [26]. Myd88-dependent IL-12 expression occurring after macrophage infection by type II strains but not type I strains was confirmed in another study [27]. Again, the involvement of Myd88 in the production of IL-12 suggested a role of Toll-like receptors (TLRs) in the immunity against T. gondii type II strains [27]. Types I and II strains of T. gondii also differentially influenced the activity of NF-κB, the transcription factor reported to play a key role in the induction of pro-inflammatory cytokine expression [28, 29]. NF-κB translocation to the nucleus was observed after the infection of mouse splenocytes or mouse bone marrow-derived macrophages (BMM) with the ME49 strain but not with the RH strain [27, 30]. The high IL-12 expression level was suggested to be specific for type II strain, as this elevated cytokine production was not inhibited by an earlier infection with a type I strain [27, 31]. ME49 induced also higher levels of IL-10, IL-1β, and IL-6 cytokines [27].
TLRs expression levels differ within placental cells
So far, no study has been performed to analyze the role of TLRs, regulators of innate immunity, in the immune response to various T. gondii strains during pregnancy and in parasite transmission through the placenta. However, several studies reported the expression of TLRs in trophoblasts, decidual cells, and amniotic epithelium [32–34]. Among them, most studies reported variable TLR expression levels observed in trophoblasts [35–37]. For TLR4, elevated expression was observed in decidual cells as compared to interstitial trophoblasts. This suggested a possibly protective role of maternally derived cells [34]. Within the first-trimester placenta, the expression of TLR2 and TLR4 was observed only in villous cytotrophoblasts and EVTs, but not in ST [35]. This suggested that placental tissue and the fetus might be infected by microbes, which have passed through the breached TLR-negative ST [38].
Taking into account the above data, which confirm the preferential colonization of EVTs by T. gondii, an important role of placental TLRs in the immune response against T. gondii seems plausible. However, previous studies showed variable expression of other TLRs in different placental cells, which seemed to be related to the cell type as well as the stage of pregnancy [39–41]. In vitro studies using cultured placental cells showed that both cytotrophoblasts and ST cells express TLR2, TLR3, TLR4, TLR5, TLR6, and TLR9 [33]. Transcription of the genes encoding TLR1, TLR2, TLR3, and TLR4, but not TLR6 was reported for first-trimester trophoblasts [35]. Expression of TLR6 observed in third-trimester trophoblasts suggested its time-dependent regulation [33, 35].
Based on current data, we hypothesize that the gestational stage-dependent differences in transplacental transmission rates of T. gondii (ranging from 0 % in case of maternal infection acquired before gestation to 67 % when infection was acquired between weeks 31 and 34 of pregnancy) might be related to the stage-dependent activity of TLRs, which is to be confirmed by further studies [42, 43]. Simultaneously, the limited expression of TLRs on trophoblast cells at the early stages of pregnancy shows that these early cells are less able to deal with intrauterine infections than differentiated trophoblasts [38]. The TLR4 expression level was elevated at term as compared to first-trimester trophoblasts [44]. We suggest that description of the mechanisms of TLRs action after primary T. gondii infection of placental cells is needed. As differential replication rates were observed after the infection of trophoblasts with various parasite strains, investigation of the role of TLRs in the course of intrauterine infection with different T. gondii strains seems to be a challenge as well.
SNPs in TLRs correlate with various infectious diseases
Single-nucleotide polymorphisms (SNPs) are common genetic alterations that may impact the expression levels of genes within which they are located. Studies of the involvement of TLR SNPs in the course of congenital toxoplasmosis seem necessary [29]. TLR polymorphisms were broadly investigated in the immune responses against various pathogens, including Hepatitis C virus (HCV), Legionella pneumophila, Plasmodium falciparum, Mycobacterium leprae, Mycobacterium tuberculosis, as well as Human cytomegalovirus (HCMV) [45–48]. Many studies demonstrated the involvement of different TLR SNPs in the course of inflammatory diseases and in the altered expression of TLR-dependent immune response genes [48–52].
The TLR4 +896 allele was highly associated with post-meningitis hearing loss, especially in case of meningococcal meningitis (MM) [53]. SNPs located in the TLR2, TLR4, and TLR9 genes were suggested as important modifications involved in immune response to BM and its clinical consequences [53]. In another study, the TLR7 Leu11Gln polymorphism was associated with Human immunodeficiency virus (HIV) disease, including higher viral load and faster progression of advanced immunosuppression [54]. In case of HCV infection, important risk factors were, among others, TLR7 IVS2 -151 G and TLR8 -129 G polymorphisms [47]. The occurrence of CD14+ cells from subjects with TLR7 IVS2 -151 A/TLR8 -129 G (AG) haplotype with the expression of TLR7 and TLR8 was significantly lower than from individuals with GG and AC haplotypes [47]. The TLR8 Met1Val polymorphism was reported to associate with both HIV and tuberculosis [55, 56]. Another study showed a slight effect of the TLR8 Met1Val allele on TNF-α response [57]. 2258 G > A SNP residing in the TLR2 gene was shown to correlate with severe phenotype in a subgroup of atopic dermatitis patients [48]. Carriers of the 2258 G > A allele showed higher risk for the development of atopy, increased levels of serum IgE and allergen-specific IgE antibodies [50]. The −16934 A > T SNP of the TLR2 gene promoter region was related to decreased risk of allergic sensitization development, hay fever, and asthma in farmer’s children [58].
In case of the TLR4 gene, the 1063 A > G and 1363 C > T SNPs were the most commonly studied. So far, these polymorphisms have been showed to associate with susceptibility to infectious diseases caused by Gram-negative bacteria, Brucella species, Respiratory syncytial virus (RSV), and P. falciparum [59–61]. Firstly, Arbour et al. reported 1063 A > G and 1363 C > T to be associated with weakened response toward inhaled lipopolysaccharide (LPS) [62]. Ducloux et al. showed a correlation of these two SNPs with rates of acute rejection and the occurrence of atherosclerotic events in kidney recipients [63]. According to the studies of Arbour et al. and Schwartz, it was shown that cells transfected with any of the TLR4 haplotypes had decreased NF-κB activity compared with normal TLR4 [62, 64–66]. A significant correlation was shown between 1063 A > G and 1363 C > T SNPs and RSV bronchiolitis in infants [61]. Few studies showed a correlation of TLR9 polymorphisms with allergy or asthma [51, 67, 68]. Peixoto-Rangel et al. reported a correlation of the 1635 A > G SNP residing in the TLR9 gene with toxoplasmic retinochoroiditis in Brazil [69]. In this population, ocular toxoplasmosis was associated with allele C at 1635 A > G (odds ratio = 7; 95 % confidence interval 1.6–30.8), which was at a frequency of 0.424, similar to that observed in European populations. The observed correlations suggested that direct interaction between T. gondii and TLR9 might trigger pro-inflammatory responses and, hence, lead to severe pathologies such as ocular disease associated with this infection in Brazil [69]. It seems important to describe the possible contribution of TLR SNPs to congenital toxoplasmosis [29].
Concluding remarks
There is a clear need for a detailed description of mechanisms of congenital toxoplasmosis development. During the maternal–fetal transmission of Toxoplasma gondii, preferential colonization of extravillous trophoblasts (EVTs) by the parasite was observed. Several studies showed different expression levels of Toll-like receptors (TLRs), dependent on placental cell type and stage of pregnancy. In case of TLR2 and TLR4, the expression was identified in first-trimester villous cytotrophoblasts and EVTs, but not in syncytiotrophoblasts (STs). Hence, an important role of TLRs against the development of fetal T. gondii infection seems plausible. SNPs located in various TLR genes associate with differential infectious diseases, including toxoplasmic retinochoroiditis. As a differential immune response has been reported in correlation with particular T. gondii genotypes, the contribution of TLRs in the course of congenital infection with various T. gondii strains seems to be likely as well. We suggest further studies of TLRs in the maternal–fetal transmission of particular parasite strains and congenital toxoplasmosis as being extremely important.
References
Delhaes L, Ajzenberg D, Sicot B, Bourgeot P, Dardé ML, Dei-Cas E, Houfflin-Debarge V (2010) Severe congenital toxoplasmosis due to a Toxoplasma gondii strain with an atypical genotype: case report and review. Prenat Diagn 30(9):902–905
Khan A, Jordan C, Muccioli C, Vallochi AL, Rizzo LV, Belfort R Jr, Vitor RW, Silveira C, Sibley LD (2006) Genetic divergence of Toxoplasma gondii strains associated with ocular toxoplasmosis, Brazil. Emerg Infect Dis 12(6):942–949
Nowakowska D, Colón I, Remington JS, Grigg M, Golab E, Wilczynski J, Sibley LD (2006) Genotyping of Toxoplasma gondii by multiplex PCR and peptide-based serological testing of samples from infants in Poland diagnosed with congenital toxoplasmosis. J Clin Microbiol 44(4):1382–1389
Rajendran C, Su C, Dubey JP (2012) Molecular genotyping of Toxoplasma gondii from Central and South America revealed high diversity within and between populations. Infect Genet Evol 12(2):359–368
Demar M, Ajzenberg D, Maubon D, Djossou F, Panchoe D, Punwasi W, Valery N, Peneau C, Daigre JL, Aznar C, Cottrelle B, Terzan L, Dardé ML, Carme B (2007) Fatal outbreak of human toxoplasmosis along the Maroni River: epidemiological, clinical, and parasitological aspects. Clin Infect Dis 45(7):e88–e95
Rico-Torres CP, Figueroa-Damián R, López-Candiani C, Macías-Avilés HA, Cedillo-Peláez C, Cañedo-Solares I, Luna-Pastén H, Tecuatl-Herrada BL, Correa D (2012) Molecular diagnosis and genotyping of cases of perinatal toxoplasmosis in Mexico. Pediatr Infect Dis J 31(4):411–413
Dardé ML (2004) Genetic analysis of the diversity in Toxoplasma gondii. Ann Ist Super Sanita 40(1):57–63
Dubey JP, Ferreira LR, Martins J, McLeod R (2012) Oral oocyst-induced mouse model of toxoplasmosis: effect of infection with Toxoplasma gondii strains of different genotypes, dose, and mouse strains (transgenic, out-bred, in-bred) on pathogenesis and mortality. Parasitology 139(1):1–13
Zenner L, Foulet A, Caudrelier Y, Darcy F, Gosselin B, Capron A, Cesbron-Delauw MF (1999) Infection with Toxoplasma gondii RH and Prugniaud strains in mice, rats and nude rats: kinetics of infection in blood and tissues related to pathology in acute and chronic infection. Pathol Res Pract 195(7):475–485
Boughattas S, Abdallah RB, Siala E, Aoun K, Bouratbine A (2011) An atypical strain associated with congenital toxoplasmosis in Tunisia. New Microbiol 34(4):413–416
Ferreira IM, Vidal JE, de Mattos CdeC, de Mattos LC, Qu D, Su C, Pereira-Chioccola VL (2011) Toxoplasma gondii isolates: multilocus RFLP-PCR genotyping from human patients in Sao Paulo State, Brazil identified distinct genotypes. Exp Parasitol 129(2):190–195
Khan A, Su C, German M, Storch GA, Clifford DB, Sibley LD (2005) Genotyping of Toxoplasma gondii strains from immunocompromised patients reveals high prevalence of type I strains. J Clin Microbiol 43(12):5881–5887
Ajzenberg D, Cogné N, Paris L, Bessières MH, Thulliez P, Filisetti D, Pelloux H, Marty P, Dardé ML (2002) Genotype of 86 Toxoplasma gondii isolates associated with human congenital toxoplasmosis, and correlation with clinical findings. J Infect Dis 186(5):684–689
Ajzenberg D, Yera H, Marty P, Paris L, Dalle F, Menotti J, Aubert D, Franck J, Bessières MH, Quinio D, Pelloux H, Delhaes L, Desbois N, Thulliez P, Robert-Gangneux F, Kauffmann-Lacroix C, Pujol S, Rabodonirina M, Bougnoux ME, Cuisenier B, Duhamel C, Duong TH, Filisetti D, Flori P, Gay-Andrieu F, Pratlong F, Nevez G, Totet A, Carme B, Bonnabau H, Dardé ML, Villena I (2009) Genotype of 88 Toxoplasma gondii isolates associated with toxoplasmosis in immunocompromised patients and correlation with clinical findings. J Infect Dis 199(8):1155–1167
Gilbert RE, Freeman K, Lago EG, Bahia-Oliveira LM, Tan HK, Wallon M, Buffolano W, Stanford MR, Petersen E; European Multicentre Study on Congenital Toxoplasmosis (EMSCOT) (2008) Ocular sequelae of congenital toxoplasmosis in Brazil compared with Europe. PLoS Negl Trop Dis 2(8):e277
Pezerico SB, Langoni H, Da Silva AV, Da Silva RC (2009) Evaluation of Toxoplasma gondii placental transmission in BALB/c mice model. Exp Parasitol 123(2):168–172
He N, Aosai F, Luo W-T, Ueda M, Yang T-H, Yamashita K, Sekiya S, Yano A (1997) Parasite load in pregnant mice infected by Toxoplasma gondii assayed by quantitative competitive-PCR. Parasitol Int 46:143–147
Loke YW (1982) Transmission of parasites across the placenta. Adv Parasitol 21:155–228
Shiono Y, Mun HS, He N, Nakazaki Y, Fang H, Furuya M, Aosai F, Yano A (2007) Maternal–fetal transmission of Toxoplasma gondii in interferon-gamma deficient pregnant mice. Parasitol Int 56(2):141–148
Robbins JR, Zeldovich VB, Poukchanski A, Boothroyd JC, Bakardjiev AI (2012) Tissue barriers of the human placenta to infection with Toxoplasma gondii. Infect Immun 80(1):418–428
Angeloni MB, Silva NM, Castro AS, Gomes AO, Silva DA, Mineo JR, Ferro EA (2009) Apoptosis and S phase of the cell cycle in BeWo trophoblastic and HeLa cells are differentially modulated by Toxoplasma gondii strain types. Placenta 30(9):785–791
Glaser KC, Hagos B, Molestina RE (2011) Effects of Toxoplasma gondii genotype and absence of host MAL/Myd88 on the temporal regulation of gene expression in infected microglial cells. Exp Parasitol 129(4):409–413
Rosowski EE, Lu D, Julien L, Rodda L, Gaiser RA, Jensen KD, Saeij JP (2011) Strain-specific activation of the NF-kappaB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. J Exp Med 208(1):195–212
Saeij JP, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC (2007) Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 445(7125):324–327
Xiao J, Jones-Brando L, Talbot CC Jr, Yolken RH (2011) Differential effects of three canonical Toxoplasma strains on gene expression in human neuroepithelial cells. Infect Immun 79(3):1363–1373
Kim L, Butcher BA, Lee CW, Uematsu S, Akira S, Denkers EY (2006) Toxoplasma gondii genotype determines MyD88-dependent signaling in infected macrophages. J Immunol 177(4):2584–2591
Robben PM, Mordue DG, Truscott SM, Takeda K, Akira S, Sibley LD (2004) Production of IL-12 by macrophages infected with Toxoplasma gondii depends on the parasite genotype. J Immunol 172(6):3686–3694
Saeij JP, Boyle JP, Boothroyd JC (2005) Differences among the three major strains of Toxoplasma gondii and their specific interactions with the infected host. Trends Parasitol 21(10):476–481
Wujcicka W, Wilczyński J, Nowakowska D (2013) SNPs in toll-like receptor (TLR) genes as new genetic alterations associated with congenital toxoplasmosis? Eur J Clin Microbiol Infect Dis 32(4):503–511
Dobbin CA, Smith NC, Johnson AM (2002) Heat shock protein 70 is a potential virulence factor in murine toxoplasma infection via immunomodulation of host NF-kappa B and nitric oxide. J Immunol 169(2):958–965
Mordue DG, Sibley LD (2003) A novel population of Gr-1+-activated macrophages induced during acute toxoplasmosis. J Leukoc Biol 74(6):1015–1025
Gillaux C, Méhats C, Vaiman D, Cabrol D, Breuiller-Fouché M (2011) Functional screening of TLRs in human amniotic epithelial cells. J Immunol 187(5):2766–2774
Mitsunari M, Yoshida S, Shoji T, Tsukihara S, Iwabe T, Harada T, Terakawa N (2006) Macrophage-activating lipopeptide-2 induces cyclooxygenase-2 and prostaglandin E(2) via toll-like receptor 2 in human placental trophoblast cells. J Reprod Immunol 72(1–2):46–59
Schatz F, Kayisli UA, Vatandaslar E, Ocak N, Guller S, Abrahams VM, Krikun G, Lockwood CJ (2012) Toll-like receptor 4 expression in decidual cells and interstitial trophoblasts across human pregnancy. Am J Reprod Immunol 68(2):146–153
Abrahams VM, Bole-Aldo P, Kim YM, Straszewski-Chavez SL, Chaiworapongsa T, Romero R, Mor G (2004) Divergent trophoblast responses to bacterial products mediated by TLRs. J Immunol 173(7):4286–4296
Holmlund U, Cebers G, Dahlfors AR, Sandstedt B, Bremme K, Ekström ES, Scheynius A (2002) Expression and regulation of the pattern recognition receptors Toll-like receptor-2 and Toll-like receptor-4 in the human placenta. Immunology 107(1):145–151
Kumazaki K, Nakayama M, Yanagihara I, Suehara N, Wada Y (2004) Immunohistochemical distribution of Toll-like receptor 4 in term and preterm human placentas from normal and complicated pregnancy including chorioamnionitis. Hum Pathol 35(1):47–54
Koga K, Mor G (2010) Toll-like receptors at the maternal–fetal interface in normal pregnancy and pregnancy disorders. Am J Reprod Immunol 63(6):587–600
Koga K, Mor G (2008) Expression and function of toll-like receptors at the maternal–fetal interface. Reprod Sci 15(3):231–242
Ma Y, Krikun G, Abrahams VM, Mor G, Guller S (2007) Cell type-specific expression and function of toll-like receptors 2 and 4 in human placenta: implications in fetal infection. Placenta 28(10):1024–1031
Nitsche JF, Jiang SW, Brost BC (2010) Toll-like receptor-2 and toll-like receptor-4 expression on maternal neutrophils during pregnancy. Am J Reprod Immunol 64(6):427–434
Nowakowska D, Wujcicka W, Sobala W, Spiewak E, Gaj Z, Wilczyński J (2013) Age-associated prevalence of Toxoplasma gondii in 8281 pregnant women in Poland between 2004 and 2012. Epidemiol Infect 1–6
Remington JS, McLeod R, Wilson CB, Desmonts G (2011) Toxoplasmosis. In: Remington JS, Klein JO, Wilson CB, Nizet V, Maldonado Y (eds) Infectious diseases of the fetus and newborn infant, 7th edn. Elsevier Saunders, Philadelphia, pp 918–1041
Beijar EC, Mallard C, Powell TL (2006) Expression and subcellular localization of TLR-4 in term and first trimester human placenta. Placenta 27(2–3):322–326
Netea MG, Wijmenga C, O’Neill LA (2012) Genetic variation in Toll-like receptors and disease susceptibility. Nat Immunol 13(6):535–542
Texereau J, Chiche JD, Taylor W, Choukroun G, Comba B, Mira JP (2005) The importance of Toll-like receptor 2 polymorphisms in severe infections. Clin Infect Dis 41(Suppl 7):S408–S415
Wang CH, Eng HL, Lin KH, Chang CH, Hsieh CA, Lin YL, Lin TM (2011) TLR7 and TLR8 gene variations and susceptibility to hepatitis C virus infection. PLoS One 6(10):e26235
Wujcicka W, Wilczyński J, Nowakowska D (2013) Alterations in TLRs as new molecular markers of congenital infections with Human cytomegalovirus? Pathog Dis. doi:10.1111/2049-632X.12083
Ahmad-Nejad P, Mrabet-Dahbi S, Breuer K, Klotz M, Werfel T, Herz U, Heeg K, Neumaier M, Renz H (2004) The toll-like receptor 2 R753Q polymorphism defines a subgroup of patients with atopic dermatitis having severe phenotype. J Allergy Clin Immunol 113(3):565–567
Kormann MS, Ferstl R, Depner M, Klopp N, Spiller S, Illig T, Vogelberg C, von Mutius E, Kirschning CJ, Kabesch M (2009) Rare TLR2 mutations reduce TLR2 receptor function and can increase atopy risk. Allergy 64(4):636–642
Lazarus R, Klimecki WT, Raby BA, Vercelli D, Palmer LJ, Kwiatkowski DJ, Silverman EK, Martinez F, Weiss ST (2003) Single-nucleotide polymorphisms in the Toll-like receptor 9 gene (TLR9): frequencies, pairwise linkage disequilibrium, and haplotypes in three U.S. ethnic groups and exploratory case–control disease association studies. Genomics 81(1):85–91
Nahum A, Dadi H, Bates A, Roifman CM (2012) The biological significance of TLR3 variant, L412F, in conferring susceptibility to cutaneous candidiasis, CMV and autoimmunity. Autoimmun Rev 11(5):341–347
van Well GT, Sanders MS, Ouburg S, van Furth AM, Morré SA (2012) Polymorphisms in Toll-like receptors 2, 4, and 9 are highly associated with hearing loss in survivors of bacterial meningitis. PLoS One 7(5):e35837
Oh DY, Baumann K, Hamouda O, Eckert JK, Neumann K, Kücherer C, Bartmeyer B, Poggensee G, Oh N, Pruss A, Jessen H, Schumann RR (2009) A frequent functional toll-like receptor 7 polymorphism is associated with accelerated HIV-1 disease progression. AIDS 23(3):297–307
Davila S, Hibberd ML, Hari Dass R, Wong HE, Sahiratmadja E, Bonnard C, Alisjahbana B, Szeszko JS, Balabanova Y, Drobniewski F, van Crevel R, van de Vosse E, Nejentsev S, Ottenhoff TH, Seielstad M (2008) Genetic association and expression studies indicate a role of toll-like receptor 8 in pulmonary tuberculosis. PLoS Genet 4(10):e1000218
Oh DY, Taube S, Hamouda O, Kücherer C, Poggensee G, Jessen H, Eckert JK, Neumann K, Storek A, Pouliot M, Borgeat P, Oh N, Schreier E, Pruss A, Hattermann K, Schumann RR (2008) A functional toll-like receptor 8 variant is associated with HIV disease restriction. J Infect Dis 198(5):701–709
Clifford HD, Yerkovich ST, Khoo SK, Zhang G, Upham J, Le Souëf PN, Richmond P, Hayden CM (2012) Toll-like receptor 7 and 8 polymorphisms: associations with functional effects and cellular and antibody responses to measles virus and vaccine. Immunogenetics 64(3):219–228
Eder W, Klimecki W, Yu L, von Mutius E, Riedler J, Braun-Fahrländer C, Nowak D, Martinez FD; ALEX Study Team (2004) Toll-like receptor 2 as a major gene for asthma in children of European farmers. J Allergy Clin Immunol 113(3):482–488
Bryant AE, Genc M, Hurtado RM, Chen KT (2004) Pulmonary Kaposi’s sarcoma in pregnancy. Am J Perinatol 21(6):355–363
Mockenhaupt FP, Hamann L, von Gaertner C, Bedu-Addo G, von Kleinsorgen C, Schumann RR, Bienzle U (2006) Common polymorphisms of toll-like receptors 4 and 9 are associated with the clinical manifestation of malaria during pregnancy. J Infect Dis 194(2):184–188
Tal G, Mandelberg A, Dalal I, Cesar K, Somekh E, Tal A, Oron A, Itskovich S, Ballin A, Houri S, Beigelman A, Lider O, Rechavi G, Amariglio N (2004) Association between common Toll-like receptor 4 mutations and severe respiratory syncytial virus disease. J Infect Dis 189(11):2057–2063
Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, Schwartz DA (2000) TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet 25(2):187–191
Ducloux D, Deschamps M, Yannaraki M, Ferrand C, Bamoulid J, Saas P, Kazory A, Chalopin JM, Tiberghien P (2005) Relevance of Toll-like receptor-4 polymorphisms in renal transplantation. Kidney Int 67(6):2454–2461
Schwartz DA (2001) The role of TLR4 in endotoxin responsiveness in humans. J Endotoxin Res 7(5):389–393
Schwartz DA (2001) Inhaled endotoxin, a risk for airway disease in some people. Respir Physiol 128(1):47–55
Schwartz DA (2002) The genetics of innate immunity. Chest 121(3 Suppl):62S–68S
Berghöfer B, Frommer T, König IR, Ziegler A, Chakraborty T, Bein G, Hackstein H (2005) Common human Toll-like receptor 9 polymorphisms and haplotypes: association with atopy and functional relevance. Clin Exp Allergy 35(9):1147–1154
Novak N, Yu CF, Bussmann C, Maintz L, Peng WM, Hart J, Hagemann T, Diaz-Lacava A, Baurecht HJ, Klopp N, Wagenpfeil S, Behrendt H, Bieber T, Ring J, Illig T, Weidinger S (2007) Putative association of a TLR9 promoter polymorphism with atopic eczema. Allergy 62(7):766–772
Peixoto-Rangel AL, Miller EN, Castellucci L, Jamieson SE, Peixe RG, Elias LdeS, Correa-Oliveira R, Bahia-Oliveira LM, Blackwell JM (2009) Candidate gene analysis of ocular toxoplasmosis in Brazil: evidence for a role for toll-like receptor 9 (TLR9). Mem Inst Oswaldo Cruz 104(8):1187–1190
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Wujcicka, W., Wilczyński, J. & Nowakowska, D. Do the placental barrier, parasite genotype and Toll-like receptor polymorphisms contribute to the course of primary infection with various Toxoplasma gondii genotypes in pregnant women?. Eur J Clin Microbiol Infect Dis 33, 703–709 (2014). https://doi.org/10.1007/s10096-013-2017-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-013-2017-3