
Abstract
The article describes paraganglioma case in woman with von Hippel–Lindau disease. She was found to be a carrier of a rare germline mutation in the VHL gene (393C>A; N131K). The patient developed large, untypical for von Hippel–Lindau disease, carotid body paraganglioma at the common carotid artery bifurcation. The carotid body paraganglioma coexisted with the haemangioblastoma situated intramedullary in region C5/C6. The haemangioblastoma reached the right-sided dorsal part of the spinal cord in section C5/C6. It produced radicular symptoms within C5/C6, followed by the later paresis of the right limbs. The haemangioblastoma was resected completely. Twelve months after the operation, the spinal symptoms receded and the carotid body paraganglioma still was asymptomatic. The current case of carotid body paraganglioma in patient with the 393C>A (N131K) missense mutation in the VHL gene, supports association of this specific mutation and VHL disease type 2, and suggests its correlation with susceptibility to paragangliomas.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Von Hippel–Lindau disease is a syndrome of high genetic predisposition towards neoplasms, which shows genealogical traits of autosomal dominant inheritance. The frequency of occurrence of this condition is estimated at 1 per 36 thousand people, which means that in Poland there are about 1,000 patients suffering from VHL disease. The condition is caused by carrying germline mutations of the VHL gene. The diagnosis of VHL disease is established based on genealogical/clinical criteria and an analysis of carrying mutations of the VHL gene. Families who are affected by VHL undergo a series of preventive/diagnostic tests and examinations whose aim is early detection and treatment of neoplastic lesions [1]. Von Hippel–Lindau disease often manifests itself with the following conditions: haemangioblastoma cerebelli, angioma of the brain stem and of the spinal cord, retinal angiomatosis, renal cysts, pancreas cysts, pheochromocytoma or renal carcinoma [2].
Paragangliomas, especially those situated extrarenally, occur very rarely in Von Hippel–Lindau disease [3–8]. They develop within the adrenal medulla, abdominal cavity, chest and occasionally—in the head and the neck. Zak and Lawson [9] quote almost 20 sites within the head and the neck where paragangliomas may occur, not only in Von Hippel–Lindau disease.
The paraganglioma in the head and the neck is connected to the parasympathetic system, while in the adrenal medulla—to the sympathetic system. In Von Hippel–Lindau disease the adrenal medulla is the place where the tumour of the pheochromocytoma type develops. Up to 5% of VHL patients die of pheochromocytoma-induced endogenous catecholamine intoxication [10].
Paragangliomas producing catecholamine lead to hypertensive crises and are thought to be the cause of various clinical symptoms of sudden onset.
Since paragangliomas of the neck occur very rarely in Von Hippel–Lindau disease, we decided to discuss the case of a patient with a lesion of this type on the neck and an haemangioblastoma of the spinal cord. The latter was treated surgically in the Department. No record has been found in Poland of a patient with Von Hippel–Lindau disease and a paraganglioma of the neck.
Case report
The female patient, aged 49, reported pains in her second and third finger of the right hand that she had had for 2 years. Initial diagnosis revealed isthmus carpalis and the patient was operated on without much success—no improvement was noticed.
A year later, intensifying root pains of Root C5 and C6 on the right side were the reason for performing an MRI scan of the cervical spine. The findings from the study showed an intramedullary lesion of 1.5 cm in diameter, localized at the height of C5–C6 with accompanying syrinx stretching from C3 to C7. The patient was consulted neurosurgically several times, but refused to submit to a surgical treatment. Gradually, the pains extended to the entire forearm of the upper right limb, and numbness appeared of both lower limbs. She met the clinical diagnostic criteria for VHL disease [11]. Sequencing of the VHL gene revealed the presence of the 393C>A (N131K) missense mutation (Fig. 1) [12]. VHL disease was also diagnosed in her relatives (shown in Fig. 2).

Sequencing chromatogram showing the 393C>A (N131K) germline mutation in the VHL gene. The mutation is marked by an arrow

Pedigree of family with VHL disease caused by the N131K mutation. Solid squares indicate affected men, solid circles indicate affected females. The type of cancer and age of diagnosis are indicated next to the symbol. A plus near the symbol indicates the presence of the mutation. A minus near the symbol indicates mutation negative cases. r hbl retinal haemangioblastoma, CNS hbl central nervous system haemangioblastoma, d48 age of death, 49 current age of a patient
The patient was treated surgically several times at the Ophthalmological Department because of gradual deterioration of vision in the left eye. Presently, the patient claims to be blind in the left eye. An ophthalmological examination proved the presence of a vast retinal angiomatosis of the left eye.
Moreover, a CT scan of the abdominal cavity showed renal cysts.
In the clinical examination carried out on admission to the Department of Neurosurgery the following conditions were found: amaurosis oculi sinistri, paraesthesiae to algetic stimuli in the medial part of the right forearm and first , second and third fingers of the right arm increased deep reflexes in the upper right limb along with decreased muscle power of this limb, as well as Babiński syndrome on the right side; RR: 134/85 mmHg; pulse: 76/min.
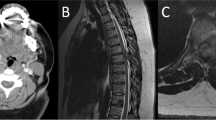
An MRI scan of the cervical spine showed a contrast enhancing image of an intramedullary lesion located near C5/C6 and extending to the dorsal part of the spinal cord on the right side to the subarachnoid space. Upwards to C3 and downwards to C7 syrinx was identified. An MRI of the head demonstrated an angioma of 1.5 cm in diameter, localized in the choroid fissure on the right side with a small cyst.
The patient was treated surgically. A laminectomy was performed from C3 to C6. Then the spinal cord was incised along the sulcus medianus posterior to uncover a lesion of the type of haemangioblastoma and a syrinx filled with yellow liquid. The neoplastic lesion was localized excentrically in the dorsal part of the spinal cord on its right side; it reached the subarachnoid space and was connected to Root C5 originating in the spinal cord. The tumour was excised completely. In the postoperative course, some paraesthesia and pains in the upper left limb appeared, which gradually subsided within 3 weeks.
After the surgery, the upper right limb pain subsided and the right-sided hemiparesis retreated. A follow-up MRI showed a total resection of the tumour.
Below we present an MRI image of the cervical spine before and after the surgery (Fig. 3a, b).

a MR of the cervical spine, sagital section: intramedullary tumour at the height of C5/C6 with syrinx reaching as far as C3 and below the lesion to C7. b MR of the cervical spine, sagital section: total resection of the tumour; a considerable reduction of the syrinx around the tumour
Findings from a histopathological test showed haemangioblastoma.
At a follow-up examination estimating how complete the excision had been, a tumour was discerned at the common carotid artery bifurcation on the right side. As a result, a detailed examination of the patient was made at the Radiodiagnostics Department of the Comprehensive Cancer Centre in Gliwice.
The radiological examination showed a carotid body paraganglioma of 45 × 32 × 24 mm3 on the right side (Fig. 4a, b)

a MR of the neck, frontal section: tumour in the carotid bifurcation, of the type of paraganglioma, on the right side. b MR of the neck, axial section: tumour in the parapharyngeal space on the right side, of the type of paraganglioma
The tumour can be seen in the right carotid space, in the right common carotid artery (CCA) bifurcation into the internal carotid artery (ICA) and the external carotid artery. The tumour appears hypointense in unenhanced T1-weighted images. “Salt and pepper” signal pattern of the tumour is visible in T2-weighted images.
The tumour appears fast and intense after contrast enhancement.
Time signal intensive curves calculated from the tumour, CCA, ICA appear to be of the same shape, maximum value of the signal intensity and maximum contrast enhancement at the same time. These signs evidence the heavy vascularization of the tumour. There are irregular no-signal spots in T2 in the lesion, which may be consistent with haemosiderin. The tumour models and translocates outwards the neighbouring vascular structures without infiltrating them; it models the parapharyngeal space and the right-sided wall of the pharynx, as well as to some extent the pterygeal muscles. The upper pole of the tumour is adjacent to the cranial basis near the carotid canal inlet, without traces of infiltration. The posterior part of the tumour leans against the I and II vertebra arches on the space of about 1.5 cm, without traces of infiltration. No traces of infiltrating the intervertebral foramen were found. The lower pole of the tumour is located about 2 cm above the division of the common carotid artery. The tumour had not given any symptoms until then. The patient refused to agree to the treatment of the carotid body paraganglioma. Laboratory testing of the noradrenaline level, adrenaline in blood and HVA in the 24-h urine collection failed to give evidence of catecholamine production by the paraganglioma.
Three weeks after the surgery of the haemangioblastoma of the spinal cord the patient was discharged from hospital in good overall condition.
Discussion
Herein we describe a rare case of carotid body paraganglioma in a woman with von Hippel–Lindau disease, caused by the N131K mutation in the VHL gene. The N131K is a rare missense mutation of the VHL gene. To date, it has been reported only once, in a family affected with VHL type 2 disease (VHL with pheochromocytoma) [13]. VHL type 2 disease is typically caused by missense mutations. However, only specific missense mutations are associated with pheochromocytoma—those mutations which map to protein binding sites of pVHL and which are predicted to cause local protein defects. The current case report of carotid body paraganglioma in patient with the 393C>A (N131K) missense mutation, further supports association of this mutation and VHL disease type 2, and suggests its correlation with susceptibility to tumours situated extrarenally. VHL kindreds with pheochromocytoma-associated missense mutations require careful surveillance for pheochromocytoma as this is frequently the first manifestation. Therefore, the correlation between the 393C>A (N131K) missense mutation and VHL type 2 may be useful in genetic counselling.
On admission to the hospital, the patient could not see in her left eye because of retinal angiomatosis; she suffered from renal cysts, haemangioblastoma near the fissura chorioidea on the right side and she had symptoms of a tumour of spinal cord. The retinal angioma found in the patient occurs in over 50% of VHL patients, often leading to blindness in the eye unless treated with laser therapy or cryocoagulation, preferably in the asymptomatic period [2].
The patient was referred to the Department of Neurosurgery because of the intramedullary tumour at C5/C6. The lesion caused pain in the upper right limb and a slight paresis of both right limbs. Pain is reported to be the most frequent symptom of haemangioblastoma of the spinal cord [14]. During the operation, it was discovered that the tumour reached the subarachnoid space of the dorsal surface of the spinal cord, and was connected to Root C5 on the right side, which may have been the reason for pains in the upper right limb. The fact that intramedullary haemangioblastomas in von Hippel–Lindau disease usually reach the dorsal surface of the medulla/spinal cord has already been reported by other authors [2, 15, 16]. The haemangioblastoma was accompanied by the syrinx much larger in size than the tumour itself, which might have been responsible for the numbness of the lower limbs. According to Wanebo et al. [17], the symptoms of VHL disease with haemangioblastoma of the spinal cord are most often caused by the syrinx and not by the tumour itself.
The tumour was not large in size—just 1.5 cm. Haemangioblastomas of the spinal cord, both the ones occurring sporadically and the ones accompanying VHL disease, are usually of small size, and the prognosis in the case of surgical treatment is favourable [15, 18]. The tumour was resected totally. After a total resection of heamangioblastoma in VHL patients, the recurrence in the same area is extremely rare [2]. One cannot exclude, however, the possibility of developing new angiomas in new locations. A histopathological examination confirmed the diagnosis of haemangioblastoma. A tumour of this type usually accompanies VHL disease [16].
In von Hippel–Lindau disease, a tumour of the medullar layer of the adrenal gland of the pheochromocytoma type occurs in 10–35% of cases [4, 10]. Lesions of the type of paraganglioma, localized extrarenally, are extremely rare [4–6, 19]. In the patient, a large lesion was found to originate in the carotid body and situated at the carotid bifurcation, on the right side. According to the radiological findings, the tumour compressed the wall of the pharynx and it displaced the neighbouring vascular structures outwards giving no clinical symptoms. The tumour is hormonally inactive; it was not joined to the cervical roots and it did not infiltrate the intervertebral foramen. In view of the absence of ailment, the patient currently refuses to submit to surgical treatment of the paraganglioma.
Oosterwijk et al. [20] report that the frequency of occurrence of surgically treated paragangliomas of the head and the neck in The Netherlands comes to one case per one million inhabitants annually. In the autopsy material, paragangliomas of the neck occur much more frequently [21, 22]. Therefore, a conclusion can be drawn that paragangliomas of the head and the neck fail to produce clinical symptoms and consequently they are not treated surgically. This is reflected in the case of the patient.
Unlike the paragangliomas of the adrenal medulla, the paragangliomas of the head and the neck usually do not show endocrinal function [3]. The case of the patient seems to confirm this observation. That is why the paragangliomas growing in the neck develop slowly, giving no clinical symptoms for a long time [3]. Incidentally, these tumours may metastasize to lungs [13].
Conclusion
The current case of carotid body paraganglioma in patient with the 393C>A (N131K) missense mutation in the VHL gene, supports association of this specific mutation with VHL disease type 2, and suggests its correlation with susceptibility to paragangliomas. VHL kindreds with pheochromocytoma-associated missense mutations require careful surveillance for pheochromocytoma as this is frequently the first manifestation. Therefore, a correlation between the 393C>A (N131K) missense mutation and VHL type 2 may be useful in genetic counselling.
References
Krzystolik K, Cybulski C, Lubiński J (1998) Choroba von Hippel–Lindau. Neur Neurochir Pol 32:1119–1133
Kongham P, Rutka T (2005) Neurocutaneous syndromes. In: Berger MS, Prados MD (eds) Textbook of neuro-oncology. Elsevier Saunders, Philadelphia, pp 826–827
Baysal BE (2002) Hereditary paraganglioma targets diverse paraganglia. J Med Genet 39:617–626
Ercolino T, Becherini L, Valeri A, Naiello M, Gagliano MS, Parenti G, Ramazzotti M, Piscitelli E, Simi L, Bergamini C, Mannelli M (2008) Uncommom clinical presentations of pheochromocytoma and paraganglioma in two different patients affected by two distinct novel germline mutations. Clin Endocrinol 68:762–768
Hull MT, Roth LM, Glover JL, Walker PD (1982) Metastatic carotid body paraganglioma in von Hippel–Lindau disease. An electron microscopic study. Arch Pathol Lab Med 106:235–239
Musso C, Paraf F, Petit B, Archanbeaud-Mouveroux F, Valleix D, Labrousse F (2000) Pancreatic neuronedocrine tumors and von Hippel-Lindau disease. Ann Pathol 20:130–133
Schimke RN, Collins DL, Rothberg PG (1998) Functioning carotid paraganglioma in the von Hippel–Lindau syndrome. Am J Med Genet 80:533–534
Zanelli M, Vanderwalt JD (1996) Carotid body paraganglioma in von Hippel–Lindau disease—a rare association. Histopathology 29:178–181
Zak FG, Lawson W (1982) Anatomy and topography. In: The paraganglionic chemoreceptor system, 1st edn. Springer-Verlag, New York, pp 15–49
Neumann H, Lips C, Hsia Y et al (1995) Von Hippel–Lindau syndrome. Brain Pathol 5:181–193
Melmon KL, Rosen SW (1964) Lindau’s disease: review of the literature and study of a large kindred. Am J Med 36:595–617
Cybulski C, Krzystolik K, Murgia A et al (2002) Germline mutations in the von Hippel–Lindau (VHL) gene in patients from Poland: disease presentation in patients with deletions of the entire VHL gene. J Med Genet 39(7):E38
Gallou C, Chauveau D, Richard S, Joly D, Giraud S, Olschwang S, Martin N, Saquet C, Chrétien Y, Méjean A, Correas JM, Benoît G, Colombeau P, Grünfeld JP, Junien C, Béroud C (2004) Genotype-phenotype correlation in von Hippel–Lindau families with renal lesions. Hum Mutat 24:215–224
Roonprapunt Ch, Silvera VM, Avi Setton, Freed D, Fred J, Jallo GI (2001) Surgical management of isolated hemangioblastomas of the spinal cord. Neurosurgery 49:321–328
Chu BC, Terae S, Hida K, Furukawa M, Abe S, Miyasaka K (2001) MR findings in spinal hemangioblastoma: correlation with symptoms and with angiographic and surgical findings. AJNR 22:206–217
Lonser RR, Weil RJ, Wanebo JE, De Vroom HL, Oldfield EH (2003) Surgical management of spinal cord hemangioblastomas in patients with von Hippel–Lindau disease. J Neurosurg 98:106–116
Wanebo JE, Lonser RR, Glenn GM, Oldfield EH (2003) The natural history of hemangioblastomas of the central nervous system in patients with von Hippel–Lindau disease. J Neurosurg 98:82–94
Van Velthoven V, Reinacher PC, Klisch JJ, Neumann HP, Glasker S (2003) Treatment of intramedullary hemangioblastomas with special attention to von Hippel–Lindau disease. Neurosurgery 53:1306–1313
Gross DJ, Avishai N, Meiner V, Filon D, Zbar B, Abeliovich D (1996) Familial pheochromocytoma associated with a novel mutation in the von Hippel–Lindau gene. J Clin Endocrinol Metab 81:147–149
Oosterwijk JC, Jansen JC, Van Schothorst EM, Oosterhof AW, Devilee P, Bakker E, Zoeteweijk MW, Van der Mey AG (1996) First experience with genetic counseling based on predictive DNA diagnosis in hereditary glomus tumours (paragangliomas). J Med Genet 33:379–383
Lack EE, Cubilla AL, Woodruff JM, Farr HW (1977) Paragangliomas of the head and neck region: a clinical study of 69 patients. Cancer 39:397–409
MacDonald RA (1956) A carotid-body-like tumor on the left subclavian artery. Arch Pathol 62:107–111
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Majchrzak, K., Cybulski, C., Bobek-Billewicz, B. et al. A case of carotid body paraganglioma and haemangioblastoma of the spinal cord in a patient with the N131K missense mutation in the VHL gene. Neurol Sci 32, 491–496 (2011). https://doi.org/10.1007/s10072-011-0502-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-011-0502-y