
Abstract
Objectives
Rheumatoid arthritis (RA) and ankylosing spondylitis (AS) have been associated with generalized and localized bone loss. We conducted a comprehensive study using imaging (dual-energy X-ray absorptiometry, DXA) and laboratory biomarkers in order to determine bone health and to study the effects of anti-tumor necrosis factor (TNF) biologics in RA and AS.
Patients and methods
Thirty-six RA and 17 AS patients undergoing 1-year etanercept (ETN) or certolizumab-pegol (CZP) therapy were studied. Bone density was assessed by DXA at baseline and after 12 months. Serum C-reactive protein (CRP), calcium, phosphate, parathyroid hormone (PTH), vitamin D3, osteocalcin, procollagen type I N-propeptide (P1NP), C-terminal telopeptide (βCTX), osteoprotegerin, sclerostin (SOST), Dickkopf-1 (DKK-1), soluble receptor activator nuclear kappa B ligand (sRANKL), and cathepsin K (cathK) levels were determined at baseline and after 6 and 12 months.
Results
TNF-α inhibition was clinically effective. Anti-TNF-α halted further bone loss over 1 year. In general, anti-TNF therapy significantly increased P1NP, SOST levels, and the P1NP/βCTX ratios, while decreased DKK-1 and CathK production at different time points in most patient subsets. In the full cohort and in RA, baseline and/or 12-month bone mineral density (BMD) at multiple sites exerted inverse relationships with CRP and βCTX, and positive correlation with SOST. In AS, L2-4 BMD after 1-year biologic therapy inversely correlated with baseline βCTX, while femoral neck BMD rather showed inverse correlations with CRP.
Conclusions
Anti-TNF therapy slowed down generalized bone loss, in association with clinical improvements, in both diseases. TNF blockade may enhance bone formation and suppress joint destruction. Anti-TNF therapy may act inversely on DKK-1 and SOST. Independent predictors of BMD were SOST and βCTX in RA, whilst CRP in AS.
Key Points • One-year anti-TNF therapy halted generalized bone loss in association with clinical improvement in arthritides. • Anti-TNF therapy may inversely act on DKK-1 and SOST. • Independent predictors of BMD were SOST and βCTX in RA, while CRP in AS. |
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) and ankylosing spondylitis (AS) have been associated with osteoporosis, as well as localized inflammatory bone resorption and/or formation [1, 2]. Low bone mineral density (BMD) and increased fracture risk have been observed in two-third of these patients [1, 2]. The RANK-RANKL system is highly involved in inflammatory bone resorption [3, 4]. Among cytokines, tumor necrosis factor α (TNF-α) induces bone loss via the stimulation of RANKL [1, 3,4,5]. TNF-α also directly induces osteoclast function [6, 7]. Interleukin-1 (IL-1), IL-6, and IL-17 also have similar effects [4]. Osteoprotegerin (OPG), a decoy receptor of RANKL, influences bone erosions in arthritides [8]. A low OPG/RANKL ratio has been associated with increased radiographic damage in RA [9, 10]. In clinical trials, TNF-α inhibitors inhibited bone destruction and reduced radiological progression in RA [11,12,13].
With respect to bone formation, wingless-related integration site (Wnt) proteins have emerged as central regulators. Upon osteoblast activation, a multi-molecular complex is formed. β-catenin becomes free, it stabilizes and it is transported to the nucleus resulting in the transcription of Wnt-induced genes regulating bone formation. This process can be inhibited by Dickkopf-1 (DKK-1) and sclerostin (SOST). DKK-1 and SOST are able to block the formation of the molecular complex mentioned above and this complex will fall into pieces [4, 14]. There is a direct interaction between DKK-1 and SOST. Blockade of DKK-1 in animal models suppresses SOST production [15]. TNF-α induces DKK-1 and DKK-1 stimulates SOST production by osteocytes. Both DKK-1 and SOST block bone formation [14]. An interaction between DKK-1 and OPG was proposed in experimental arthritis models suggesting that the antiresorptive and bone formation-inducing property of DKK-1 inhibition also involves OPG [16]. As TNF-α induces DKK-1 and SOST and thus inhibits bone formation, in contrast to RA, there have been lots of controversies with regard to the efficacy of TNF-α inhibitors in halting bone formation and the development of syndesmophytes in axial AS [12, 14]. In animal models of arthritis, SOST promoted, while anti-SOST attenuated the formation of erosions [17]. In contrast, another study suggested that SOST may act differently by inhibiting rather than promoting TNF-α-mediated inflammatory bone loss [18]. SOST may exert diverse effects on bone turnover under inflammatory and non-inflammatory conditions.
In addition to their effects on localized bone turnover, anti-TNF therapies also influence the development and progression of generalized osteoporosis in conjunction with RA and AS [12].
Anti-TNF therapies may also influence bone turnover and the production of bone biomarkers. Anti-TNF-α agents were shown to increase serum osteocalcin (OC) and procollagen type I N-propeptide (P1NP) levels, which are markers of bone formation, and suppressed C-terminal telopeptide (βCTX) and receptor activator nuclear kappa B ligand (RANKL) levels, which are markers of bone resorption, in RA and AS (reviewed in [12]). These findings were not always consistent as some studies with TNF-α inhibitors showed no effects on OC, P1NP and βCTX levels [19, 20]. On the other hand, anti-TNF agents increased OPG/RANKL, OC/βCTX, and P1NP/βCTX ratios [20,21,22]. Furthermore, TNF-α inhibitors also suppress DKK-1 leading to increased bone formation in RA [23, 24]. Interestingly, in one study, anti-TNF treatment increased SOST production in RA [25]. In most studies, changes in bone biomarkers by anti-TNF therapy were associated with improvements in disease activity and inflammatory markers (e.g. CRP) in RA and AS [12].
As most studies in the literature have been conducted in either RA or AS/SpA, here, we conducted a comprehensive study on the effects of anti-TNF biologics on bone density and bone markers in both RA and AS. Moreover, we assessed more than 10 bone markers in order to elucidate the complex effects of biologics on bone metabolism. Our aim was to draw a complex picture of, as well as to demonstrate the effects of anti-TNF therapy on bone health in RA and AS.
Patients and methods
Patients
Fifty-three patients with inflammatory arthritis (36 RA and 17 AS) were enrolled in the study. Patient characteristics are seen in Table 1. The cohort included 34 women and 19 men with mean age of 52.0 ± 12.1 (range 24–83) years. Mean disease duration was 8.5 ± 7.9 (range 1–44) years. Patients with active disease were recruited prior to initiating a biological therapy. Inclusion criteria included definitive diagnosis of RA or AS; high disease activity (DAS28 > 5.1, BASDAI > 4 after at least 3 months of combined conventional DMARD therapy); clinical indication of biological therapy. Exclusion criteria included acute/recent infection, standard contraindications to anti-TNF therapy, chronic renal or liver failure. None of the patients had known primary osteoporosis prior to the diagnosis of RA or AS. None of the patients received replacement vitamin D therapy at the time of inclusion. At baseline, RA patients had a mean DAS28 of 5.00 ± 0.86, while AS patients exerted mean BASDAI of 5.79 ± 1.19. All patients were biologic-naïve, started on an anti-TNF therapy at baseline and received the same biological treatment at 1 year. Clinical assessments were performed at baseline, and after 3, 6, and 12 months of therapy. Among the 36 RA patients, 20 received etanercept (ETN) 50 mg/week subcutaneous (SC) and 16 received certolizumab pegol (CZP) (400 mg at 0, 2, and 4 weeks, and thererafter 200 mg twice weekly SC). Altogether 28 RA patients were administered the TNF inhibitor in combination with methotrexate (MTX). These patients had been on MTX prior to the initiation of biologics and the MTX dose was not changed. All 17 AS patients received ETN 50 mg/week SC in monotherapy. Although most RA patients and some AS patients may have received corticosteroids prior to the study, none of the patients were on corticosteroids for at least 3 months prior to and during the study. The study was approved by the Hungarian Scientific Research Council Ethical Committee (approval no. 14804-2/2011/EKU). Written informed consent was obtained from each patient and assessments were carried out according to the declaration of Helsinki.
Bone densitometry assessments
Dual energy X-ray absorptiometry examination was performed using the LUNAR Prodigy (GE-Lunar Corp., Madison, WI, USA) densitometer by a single technician during the study period. The coefficient of variation (CV) of the technique at our institute was 0.8% using the anatomical spine phantom measured daily and there was absence of machine drift during the study. The short-term in vivo precision error for L2-L4 lumbar spine is 0.012 g/cm2 (LSC = 0.034 g/cm2 at 95% confidence level) and femur neck is 0.013 g/cm2 (LSC = 0.035 g/cm2 at 95% confidence level).
Laboratory measurements and assessment of disease activity
Blood was drawn after overnight fasting. Serum high sensitivity C reactive protein (hsCRP; normal, ≤ 5 mg/l) and IgM rheumatoid factor (RF; normal, ≤ 50 IU/ml) were measured by quantitative nephelometry (Cobas Mira Plus-Roche), using CRP and RF reagents (both Dialab). ACPA (anti-CCP) autoantibodies were detected in serum samples using a second-generation Immunoscan-RA CCP2 ELISA test (Euro Diagnostica; normal, ≤ 25 IU/ml). The assay was performed according to the manufacturer’s instructions. Disease activity of RA and AS was calculated as DAS28-ESR (3 variables) and BASDAI, respectively [26].
Bone biomarkers
Blood was drawn after overnight fasting. Serum calcium (Ca; Roche Diagnostics; normal, 2.1–2.6 mmol/l) and phosphate (P; Roche Diagnostics; normal, 0.8–1.45 mmol/l); parathyroid hormone (PTH; Roche Diagnostics; normal, 1.6–6.9 pmol/l); 25-hydroxy-vitamin D3 (DiaSorin; normal, ≥ 75 nmol/l); osteocalcin (OC; Roche Diagnostics; normal, < 41 μg/l), P1NP (Roche Diagnostics; normal, < 75 μg/l), βCTX (Roche Diagnostics; normal, < 0.57 μg/l), osteoprotegerin (OPG; Biomedica; median, 2.7 pmol/l), sclerostin (SOST; Biomedica; median, 24.14 pmol/l), DKK-1 (Biomedica; median, 36 pmol/l), soluble RANKL (Ampli-sRANKL; Biomedica; median, 0.14 pmol/l), and cathepsin K (cathK; Biomedica; median, 8.7 pmol/l) were determined by ELISA at baseline, 6 and 12 months after treatment initiation. The above-mentioned reference values for biochemical data were generated by the local laboratory.
Statistical analysis
Statistical analysis was performed using SPSS (IBM 22 software). Data are expressed as the mean ± SD for continuous variables and percentages for categorical variables. Continuous variables were evaluated by paired two-tailed t test and the Wilcoxon test. Nominal variables were compared between groups using the chi-squared or Fisher’s exact test, as appropriate. Correlations were determined by Spearman’s analysis. Univariate and multiple regression analysis using the stepwise method was applied to investigate independent associations between DXA measurements (dependent variables) and laboratory parameters (independent variables). Sex and menopausal status were included in the regression analyses. The β standardized linear coefficients showing linear correlations between two parameters were determined. The B (+ 95% CI) regression coefficient indicated independent associations between dependent and independent variables during changes. P values < 0.05 were considered significant.
Results
Clinical response to anti-TNF therapy in RA and AS
TNF-α inhibition was highly effective in RA and AS patients. In the RA cohort (n = 36), ETN and CZP treatment resulted in significant decreases in DAS28 after 3 months (3.52 ± 0.79; p < 0.001), 6 months (3.13 ± 0.84; p < 0.001) and 12 months of treatment (3.02 ± 0.96; p < 0.001) compared with baseline (5.00 ± 0.86) (data not shown). In RA, CRP at baseline (16.0 ± 19.1 mg/l) was also significantly higher than after 3 months (8.5 ± 11.3 mg/l; p < 0.001), 6 months (7.0 ± 7.1 mg/l; p = 0.005) and 12 months (7.5 ± 7.9 mg/l; p = 0.011) (data not shown).
In AS (n = 17), BASDAI significantly decreased from 5.79 ± 1.19 at baseline to 2.04 ± 0.89 (p < 0.001), 2.00 ± 1.03 (p < 0.001), and 1.86 ± 1.04 (p < 0.001) at 3, 6, and 12 months, respectively (data not shown). Moreover, CRP was also higher at baseline (12.5 ± 12.0 mg/l) than after 3 months (5.7 ± 13.6 mg/l; p = 0.026), 6 months (6.3 ± 13.5 mg/l; p = 0.041), and 12 months of therapy (4.4 ± 6.6 mg/l; p = 0.003) (data not shown).
Fortunately, all RA and AS patients were responders and no biologic switch was necessary in any of these patients during the 1-year treatment period.
Effects of anti-TNF therapy on bone mineral density
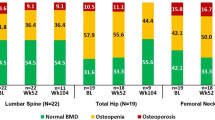
While anti-TNF-α treatment halted further generalized bone loss over 1 year in the whole cohort, there was no significant difference in mean L2–4 vertebral and femoral neck BMD or T-scores at baseline compared with 12 months (L2–4 vertebral BMD 0.890 ± 0.027 vs 0.889 ± 0.025 g/cm2 and T-score − 0.51 ± 0.21 vs − 0.46 ± 0.21; femoral neck BMD 0.842 ± 0.020 vs 0.838 ± 0.020 g/cm2 and T-score − 0.84 ± 0.17 vs − 0.89 ± 0.17, respectively). Similar observations were made in the RA and AS subsets (data not shown).
Effects of biologics on bone biomarkers
Among the bone turnover (formation and resorption) markers, in the full RA+AS cohort, one-year anti-TNF therapy significantly increased P1NP after 3 months (51.8 ± 22.5 μg/l; p = 0.042) and 6 months (53.5 ± 27.1 μg/l; p = 0.040) compared with baseline (46.7 ± 19.3 μg/l) (Fig. 1). Similar differences in P1NP levels could be observed in the RA subset after 3 months (52.2 ± 24.0 μg/l; p = 0.041) and 6 months (56.4 ± 29.0 μg/l; p = 0.026) compared with baseline (45.6 ± 19.9 μg/l) (Fig. 1). In AS, P1NP levels significantly increased after 12 months (56.9 ± 28.8 μg/l; p = 0.035) versus baseline (49.2 ± 18.4 μg/l) (Fig. 1). At the same time, OC and βCTX had normal levels at all time points. Neither OC (baseline, 20.3 ± 8.8 μg/l; 12 months, 20.5 ± 9.8 μg/l) nor βCTX (baseline, 0.34 ± 0.18 μg/l; 12 months, 0.35 ± 0.18 μg/l) changed significantly in the full RA+AS cohort.

Effects of 1-year anti-TNF therapy on P1NP production. TNF inhibition significantly increased P1NP levels in the RA+AS cohort after 3 and 6 months of treatment; in RA patients after 3 and 6 months and in AS, after 12 months
With respect to the Wnt-β-catenin pathway and its regulators, in the RA+AS cohort, DKK-1 levels were higher at baseline (59.7 ± 28.6 pmol/l) compared with the reference value described above. DKK-1 levels significantly decreased after 6 months of treatment (51.6 ± 25.5 pmol/l; p = 0.045) compared with baseline (Fig. 2A). SOST levels were also higher at baseline (94.6 ± 45.3 pmol/l) compared with the normal reference value. Conversely to DKK-1, SOST levels significantly increased after 12 months (112.4 ± 76.1 pmol/l; p = 0.035) versus baseline (Fig. 2B). In RA, DKK-1 also significantly decreased after 6 months (52.1 ± 26.1 pmol/l; p = 0.042) versus baseline (60.6 ± 28.9 pmol/l) (Fig. 2A). On the other hand, SOST significantly increased after 12 months (128.5 ± 83.7 pmol/l; p = 0.038) versus baseline (107.0 ± 47.5 pmol/l) (Fig. 2B). In AS, SOST significantly increased after 12 months (81.3 ± 46.9 pmol/l; p = 0.034) compared with baseline (70.6 ± 29.0 pmol/l) (Fig. 2B).

Effects of anti-TNF therapy on DKK-1 and SOST levels. Anti-TNF treatment significantly decreased DKK-1 production in the full cohort and in the RA subset after 6 months (A). TNF inhibition also increased SOST levels in the RA+AS, RA, and AS subsets after 12 months (B)
In the full cohort, the baseline concentrations of CathK (27.4 ± 6.8 pmol/l) were higher compared with the normal reference value above. There was a significant decrease in CathK production after 12 months (25.8 ± 5.5 pmol/l; p = 0.006) compared with baseline (Fig. 3). In RA, CathK also significantly decreased after 12 months (26.9 ± 5.6 pmol/l; p = 0.012) versus baseline (28.7 ± 6.2 pmol/l) (Fig. 3).

Effects of 1-year anti-TNF therapy on cathepsin K (CathK) production. TNF inhibition significantly decreased CathK levels in the RA+AS cohort and in RA patients after 12 months of treatment
With respect to the RANKL pathway, in the full cohort or in RA or AS patients, sRANKL and OPG levels did not show any differences during anti-TNF therapy. In addition, Ca, P, vitamin D3 and PTH levels also did not change during the 1-year period (data not shown).
When calculating bone formation/resorption ratios (P1NP/βCTX, OC/βCTX and OPG/RANKL), which better reflect bone turnover changes, in the full cohort, the P1NP/βCTX ratio significantly increased at 6 months (187.5 ± 85.5; p = 0.032) compared with baseline (160.8 ± 56.5) (Fig. 4). In RA, the P1NP/βCTX ratio significantly increased after 6 months (190.2 ± 87.2; p = 0.035) (Fig. 4). In addition, the OC/βCTX and OPG/RANKL ratios did not change significantly during the 12 months in the full cohort, RA or AS (data not shown).

Effects of 1-year anti-TNF therapy on P1NP/βCTX ratios. TNF inhibition significantly increased P1NP/βCTX in the RA+AS cohort and in RA patients after 6 months of treatment
Correlations between bone mineral density and laboratory biomarkers
Univariate and multiple regression analyses were performed in order to determine associations between BMD at various sites (dependent variables) and laboratory parameters (independent variables) (Table 2). In the full cohort and in RA patients, baseline and/or 12-month BMD at multiple sites exerted inverse relationships with CRP and βCTX, as well as positive correlations with SOST. In the AS subset, L2–4 BMD after 1-year biologic therapy inversely correlated with baseline βCTX. Femoral neck BMD rather showed inverse correlations with CRP. The actual data and p values are included in Table 2.
Discussion
RA and AS have been associated with secondary osteoporosis and increased fracture risk [1, 2]. Inflammatory bone formation is also a hallmark of AS [12, 14]. Anti-TNF biologics may inhibit bone loss, but they exert limited effects on inflammatory bone formation [12]. Furthermore, biological therapy may affect the production of bone markers in RA and AS [12]. With respect to mode of action, TNF inhibitors decrease RANKL and increase OPG expression in inflammatory diseases [12]. TNF inhibition also results in blockade of DKK-1 and stimulation of Wnt-dependent bone formation [12].
In this study, we assessed the effects of ETN or CZP therapy on disease activity, bone loss and bone biomarkers in RA and AS. As expected, 1-year anti-TNF treatment was effective in both RA and AS patients as it significantly decreased DAS28 and BASDAI, respectively. These effects of ETN and CZP were observed as early as after 3 months. The anti-inflammatory effects of anti-TNF therapy were accompanied by inhibition of bone loss in both diseases. L2–4 and femoral neck BMD remained unchanged after 1-year ETN or CZP therapy in the entire cohort, as well as in RA and AS. These effects have previously been reported by others [12, 13]. Baseline CRP inversely correlated not only with baseline L2–4 and femoral neck BMD, but also with BMD after 1-year treatment. Thus, at baseline, high-grade inflammation correlated with low BMD. Moreover, we found that baseline CRP may predict further bone loss after 1-year anti-TNF therapy.
The first publications on the effects of TNF inhibitors on bone and osteoporosis were published in the 2000s. In most studies, effects on the bone have been associated with favorable clinical responses. A great amount of data have been published on infliximab, ETN, adalimumab and some data also on golimumab (reviewed in [12]). We have not found any reports on the bone effects of CZP.
Regarding bone biomarkers, 1-year anti-TNF therapy significantly increased P1NP over time in our full cohort, in RA and AS. Other markers of bone turnover, such as OC or βCTX did not change upon anti-TNF therapy. The P1NP/βCTX ratio increased over time in the full cohort and in RA. Bone resorbing activity as indicated by βCTX at baseline was associated with low baseline BMD and we showed that baseline βCTX may also predict further bone loss after 1-year anti-TNF therapy. In line with our findings, previous studies found that TNF inhibitors increased serum P1NP levels and, in some cohorts, suppressed βCTX levels in RA and AS [20, 21, 23, 27,28,29,30,31,32]. These findings were not always consistent as some other studies showed no effects of TNF-α inhibitors on OC, P1NP, and βCTX levels [19, 20]. Furthermore, anti-TNF agents have been shown to increase OPG/RANKL, OC/βCTX, and P1NP/βCTX ratios [20,21,22]. In most studies, changes in bone biomarkers by anti-TNF therapy were associated with improvements in disease activity in RA and AS [12].
With respect to DKK-1 and SOST, upon 1-year ETN or CZP treatment, DKK-1 levels transiently decreased in the whole cohort and in the RA subset. On the other hand, SOST levels significantly increased in the entire cohort, as well as in the RA and AS subsets after 12 months of treatment. When comparing RA and AS, SOST levels were lower at all time points in AS compared with RA. Baseline SOST correlated with L2–4 and femoral neck BMD at baseline, as well as after 12 months in the whole cohort and in RA. This association was further confirmed by regression analyses. Interestingly, higher baseline SOST was associated with higher BMD at all sites. Moreover, high baseline SOST predicted higher BMD after 12 months of anti-TNF treatment. Similar to our observations, other studies in RA also reported that TNF-α inhibitors suppressed DKK-1 leading to increased bone formation [23, 24]. In addition, in one study, anti-TNF treatment increased SOST production in RA [25]. The seemingly controversial effects of anti-TNF biologics on DKK-1 and SOST need further discussion. In general, DKK-1 and SOST were thought to have unidirectional effects on the bone. Both molecules were described to block Wnt-β-catenin-dependent bone formation. TNF-α may induce both DKK-1 and SOST production [14]. DKK-1 may directly drive SOST expression by osteocytes and DKK-1 blockade also inhibits SOST production [14, 15]. Indeed, Chen et al. [17] administered anti-SOST antibody to human TNF transgenic mice that develop inflammatory arthritis. In this model, anti-SOST did not affect synovitis. However, anti-SOST, but not anti-TNF antibodies halted the progression of bone erosions [17]. On the other hand, more recently, Wehmeyer et al. [18] reported that in a TNF-α-dependent murine arthritis model, where synovial fibroblasts abundantly produce SOST, SOST inhibition leads to accelerated synovitis and joint destruction. SOST blocked TNF-α-dependent signaling pathways and SOST inhibition ameliorated arthritis in an animal model [18]. Moreover, low SOST and DKK-1 production have been associated with syndesmophyte formation in AS [33]. SOST levels remain low after TNF-α blockade, which may partly explain the inefficacy of TNF inhibitors on bone formation in AS [34, 35]. Interestingly, low SOST and DKK-1 levels in AS result in increased CRP production [35]. Indeed, SOST levels increased in our study in both AS and RA; however, the absolute serum concentrations of SOST were lower in AS vs RA at all time points. Our findings that TNF-α inhibitors may differentially regulate DKK-1 and SOST suggest that, apart from the direct stimulatory effect of DKK-1 on SOST [14, 15], there may be other indirect regulatory mechanisms between these two molecules. Finally, in our cohort, high SOST correlated with high BMD at all sites. This finding supports previous observations that, while under non-inflammatory conditions, such as osteoporosis, SOST inhibits Wnt-mediated bone formation and contributes to bone loss and joint destruction [18, 36, 37], under TNF-dependent inflammatory conditions, SOST may preserve bone [18] and attenuate joint damage [18]. Furthermore, anti-TNF treatment may increase SOST levels [25] and, as we demonstrated in this study, high SOST may correlate with high BMD.
When comparing bone resorption indicated by βCTX with bone formation associated with SOST, only βCTX, but not OC, and only SOST, but not DKK-1 correlated with BMD. Thus, our data suggest that βCTX and SOST, in addition to CRP, may be the most important biomarkers and independent predictors of BMD. Moreover, most of these associations were seen in the entire cohort, where two-third of the patients had RA, and in the RA subset alone. Interestingly only βCTX and SOST, but not CRP associated with BMD in RA. In contrast, in our relatively small AS subset, only CRP, but not βCTX or SOST correlated with femoral neck BMD. In conclusion, in RA, SOST and βCTX, but not CRP are independent predictors of BMD (at baseline and after 12 months), whilst, in AS, CRP may serve as the best biomarker of inflammatory bone metabolism. As mentioned above, low SOST and DKK-1 levels in AS may stimulate CRP production [35], this may further explain the outstanding importance of CRP in AS-related bone changes.
CathK is a matrix-degrading enzyme that effectively digests collagen. CathK has been implicated in osteoporosis and a CathK inhibitor, odanacatib, has been tried in postmenopausal osteoporosis [38]. In RA, increased CathK levels correlated with joint damage [39]. CathK is strongly expressed at different spinal regions in AS [40]. In our entire cohort and in the RA subset, but not in AS, TNF inhibition resulted in significantly decreased CathK levels after 1 year. In a study of 13 AS patients, infliximab, ETN, or adalimumab treatment did not change CathK levels [41]. We did not find any reports in the literature on the effects of biologics on CathK production.
Our study has advantages and limitations. We included RA and AS patients and compared the two disease. To our knowledge, this is the first study that assesses the effects of CZP on bone loss. Moreover, we determined a high number of bone biomarkers in these patients and correlated inflammatory and bone biomarkers with BMD. Finally, we developed a potential explanation on the differing effects of SOST in inflammatory vs non-inflammatory bone loss. This study certainly has limitations, primarily the relatively low number of patients. However, we assessed BMD, as well as a great number of biomarkers in a prospective manner, which would have been more difficult in a larger patient cohort.
In summary, despite its diverse effects on bone formation in RA and AS, anti-TNF therapy (ETN and CZP) slowed down bone loss in both diseases. Independent predictors of BMD were SOST and βCTX in RA, whilst CRP in AS. Further studies are needed to evaluate the potential beneficial effects of biologics on inflammatory bone loss.
References
Deal C (2012) Bone loss in rheumatoid arthritis: systemic, periarticular, and focal. Curr Rheumatol Rep 14(3):231–237
Magrey M, Khan MA (2010) Osteoporosis in ankylosing spondylitis. Curr Rheumatol Rep 12(5):332–336
Schett G, Hayer S, Zwerina J, Redlich K, Smolen JS (2005) Mechanisms of disease: the link between RANKL and arthritic bone disease. Nat Clin Pract Rheumatol 1(1):47–54
Takayanagi H (2009) Osteoimmunology and the effects of the immune system on bone. Nat Rev Rheumatol 5(12):667–676
Daoussis D, Andonopoulos AP, Liossis SN (2010) Wnt pathway and IL-17: novel regulators of joint remodeling in rheumatic diseases. Looking beyond the RANK-RANKL-OPG axis. Semin Arthritis Rheum 39(15):369–383
Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, Nakagawa N, Kinosaki M, Yamaguchi K, Shima N, Yasuda H, Morinaga T, Higashio K, Martin TJ, Suda T (2000) Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med 191(2):275–286
Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL (2000) TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest 106(12):1481–1488
Schett G, Redlich K, Smolen JS (2003) The role of osteoprotegerin in arthritis. Arthritis Res Ther 5(5):239–245
van Tuyl LH, Voskuyl AE, Boers M, Geusens P, Landewe RB, Dijkmans BA et al (2010) Baseline RANKL:OPG ratio and markers of bone and cartilage degradation predict annual radiological progression over 11 years in rheumatoid arthritis. Ann Rheum Dis 69(9):1623–1628
Geusens PP, Landewe RB, Garnero P, Chen D, Dunstan CR, Lems WF et al (2006) The ratio of circulating osteoprotegerin to RANKL in early rheumatoid arthritis predicts later joint destruction. Arthritis Rheum 54(6):1772–1777
Smolen JS, Landewe R, Bijlsma J, Burmester G, Chatzidionysiou K, Dougados M et al (2017) EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis 76(6):960–977
Szentpetery A, Horvath A, Gulyas K, Petho Z, Bhattoa HP, Szanto S et al (2017) Effects of targeted therapies on the bone in arthritides. Autoimmun Rev 16(3):313–320
Haugeberg G, Helgetveit KB, Forre O, Garen T, Sommerseth H, Proven A (2014) Generalized bone loss in early rheumatoid arthritis patients followed for ten years in the biologic treatment era. BMC Musculoskelet Disord 15:289
Schett G, Gravallese E (2012) Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nat Rev Rheumatol 8(11):656–664
Heiland GR, Zwerina K, Baum W, Kireva T, Distler JH, Grisanti M, Asuncion F, Li X, Ominsky M, Richards W, Schett G, Zwerina J (2010) Neutralisation of Dkk-1 protects from systemic bone loss during inflammation and reduces sclerostin expression. Ann Rheum Dis 69(12):2152–2159
Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, Dwyer D, Korb A, Smolen J, Hoffmann M, Scheinecker C, van der Heide D, Landewe R, Lacey D, Richards WG, Schett G (2007) Dickkopf-1 is a master regulator of joint remodeling. Nat Med 13(2):156–163
Chen XX, Baum W, Dwyer D, Stock M, Schwabe K, Ke HZ, Stolina M, Schett G, Bozec A (2013) Sclerostin inhibition reverses systemic, periarticular and local bone loss in arthritis. Ann Rheum Dis 72(10):1732–1736
Wehmeyer C, Frank S, Beckmann D, Bottcher M, Cromme C, Konig U et al (2016) Sclerostin inhibition promotes TNF-dependent inflammatory joint destruction. Sci Transl Med 8(330):330ra35
Marotte H, Pallot-Prades B, Grange L, Gaudin P, Alexandre C, Miossec P (2007) A 1-year case-control study in patients with rheumatoid arthritis indicates prevention of loss of bone mineral density in both responders and nonresponders to infliximab. Arthritis Res Ther 9(3):R61
Chopin F, Garnero P, le Henanff A, Debiais F, Daragon A, Roux C, Sany J, Wendling D, Zarnitsky C, Ravaud P, Thomas T (2008) Long-term effects of infliximab on bone and cartilage turnover markers in patients with rheumatoid arthritis. Ann Rheum Dis 67(3):353–357
Vis M, Havaardsholm EA, Haugeberg G, Uhlig T, Voskuyl AE, van de Stadt RJ, Dijkmans BAC, Woolf AD, Kvien TK, Lems WF (2006) Evaluation of bone mineral density, bone metabolism, osteoprotegerin and receptor activator of the NF kappa B ligand serum levels during treatment with infliximab in patients with rheumatoid arthritis. Ann Rheum Dis 65(11):1495–1499
Woo JH, Lee HJ, Sung IH, Kim TH (2007) Changes of clinical response and bone biochemical markers in patients with ankylosing spondylitis taking etanercept. J Rheumatol 34(8):1753–1759
Wang SY, Liu YY, Ye H, Guo JP, Li R, Liu X et al (2011) Circulating Dickkopf-1 is correlated with bone erosion and inflammation in rheumatoid arthritis. J Rheumatol 38(5):821–827
Garnero P, Tabassi NC, Voorzanger-Rousselot N (2008) Circulating dickkopf-1 and radiological progression in patients with early rheumatoid arthritis treated with etanercept. J Rheumatol 35(12):2313–2315
Lim MJ, Kwon SR, Joo K, Son MJ, Park SG, Park W (2014) Early effects of tumor necrosis factor inhibition on bone homeostasis after soluble tumor necrosis factor receptor use. Korean J Intern Med 29(6):807–813
Prevoo ML, van’t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL (1995) Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum 38(1):44–48
Torikai E, Kageyama Y, Takahashi M, Suzuki M, Ichikawa T, Nagafusa T, Nagano A (2006) The effect of infliximab on bone metabolism markers in patients with rheumatoid arthritis. Rheumatology (Oxford) 45(6):761–764
Visvanathan S, van der Heijde D, Deodhar A, Wagner C, Baker DG, Han J, Braun J (2009) Effects of infliximab on markers of inflammation and bone turnover and associations with bone mineral density in patients with ankylosing spondylitis. Ann Rheum Dis 68(2):175–182
Visvanathan S, Wagner C, Marini JC, Baker D, Gathany T, Han J et al (2008) Inflammatory biomarkers, disease activity and spinal disease measures in patients with ankylosing spondylitis after treatment with infliximab. Ann Rheum Dis 67(4):511–517
Barnabe C, Hanley DA (2009) Effect of tumor necrosis factor alpha inhibition on bone density and turnover markers in patients with rheumatoid arthritis and spondyloarthropathy. Semin Arthritis Rheum 39(2):116–122
Vis M, Wolbink GJ, Lodder MC, Kostense PJ, van de Stadt RJ, de Koning MH et al (2003) Early changes in bone metabolism in rheumatoid arthritis patients treated with infliximab. Arthritis Rheum 48(10):2996–2997
Seriolo B, Paolino S, Sulli A, Ferretti V, Cutolo M (2006) Bone metabolism changes during anti-TNF-alpha therapy in patients with active rheumatoid arthritis. Ann N Y Acad Sci 1069:420–427
Heiland GR, Appel H, Poddubnyy D, Zwerina J, Hueber A, Haibel H, Baraliakos X, Listing J, Rudwaleit M, Schett G, Sieper J (2012) High level of functional dickkopf-1 predicts protection from syndesmophyte formation in patients with ankylosing spondylitis. Ann Rheum Dis 71(4):572–574
Saad CG, Ribeiro AC, Moraes JC, Takayama L, Goncalves CR, Rodrigues MB et al (2012) Low sclerostin levels: a predictive marker of persistent inflammation in ankylosing spondylitis during anti-tumor necrosis factor therapy? Arthritis Res Ther 14(5):R216
Klingberg E, Nurkkala M, Carlsten H, Forsblad-d'Elia H (2014) Biomarkers of bone metabolism in ankylosing spondylitis in relation to osteoproliferation and osteoporosis. J Rheumatol 41(7):1349–1356
Piters E, Boudin E, Van Hul W (2008) Wnt signaling: a win for bone. Arch Biochem Biophys 473(2):112–116
Lerner UH, Ohlsson C (2015) The WNT system: background and its role in bone. J Intern Med 277(6):630–649
Perez-Castrillon JL, Pinacho F, De Luis D, Lopez-Menendez M, Duenas LA (2010) Odanacatib, a new drug for the treatment of osteoporosis: review of the results in postmenopausal women. J Osteoporos 2010:1–5
Skoumal M, Haberhauer G, Kolarz G, Hawa G, Woloszczuk W, Klingler A (2005) Serum cathepsin K levels of patients with longstanding rheumatoid arthritis: correlation with radiological destruction. Arthritis Res Ther 7(1):R65–R70
Neidhart M, Baraliakos X, Seemayer C, Zelder C, Gay RE, Michel BA, Boehm H, Gay S, Braun J (2009) Expression of cathepsin K and matrix metalloproteinase 1 indicate persistent osteodestructive activity in long-standing ankylosing spondylitis. Ann Rheum Dis 68(8):1334–1339
Wendling D, Cedoz JP, Racadot E (2008) Serum levels of MMP-3 and cathepsin K in patients with ankylosing spondylitis: effect of TNF alpha antagonist therapy. Joint Bone Spine 75(5):559–562
Funding
Open access funding provided by University of Debrecen (DE). This research was supported by the Hungarian National Scientific Research Fund (OTKA) grant No. K 105073 (H.P.B. and Z.S.); by the European Union and the State of Hungary and co-financed by the European Social Fund in the framework of TAMOP-4.2.4.A/2-11/1-2012-0001 ‘National Excellence Program’(Z.S.); and by the European Union grants GINOP-2.3.2-15-2016-00015 and GINOP-2.3.2-15-2016-00050 (Z.S.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures
None.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gulyás, K., Horváth, Á., Végh, E. et al. Effects of 1-year anti-TNF-α therapies on bone mineral density and bone biomarkers in rheumatoid arthritis and ankylosing spondylitis. Clin Rheumatol 39, 167–175 (2020). https://doi.org/10.1007/s10067-019-04771-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-019-04771-3