
Abstract
IgG4-related disease (IgG4-RD) and Castleman’s disease (CD) share similar clinical manifestations. When the histopathology coincides with the diagnosis of both IgG4-RD and CD, it is hard to depart the two disease entities utterly; here we call it IgG4-CD provisionally. In this study, we aim to review the clinical features of IgG4-CD. This study is based on a retrospective analysis of a prospectively acquired database. IgG4-CD was defined histopathologically in patients who fulfilled the diagnosis of both IgG4-RD and CD. Forty-five definite IgG4-RD and 16 multicentric CD (MCD) patients were recruited as controls. Clinical features including organ involvement, serum IgG4, IgG, IgE, ESR, CRP, and IL-6 levels were collected and analyzed. Fifteen patients (2.8%) out of 534 patients with IgG4-RD in China’s largest prospective IgG4-RD and Mimicry cohort fulfilled the definition of IgG4-CD. There were 14 males and 1 female, whose mean age was 47 ± 18 years old, and the median disease duration before diagnosis was 12 (1–132) months. Eight patients have allergic disease history. IgG4-CD patients had more lymph node involvement (100 vs 57.8%, P < 0.01), while IgG4-RD patients had more submandibular (33.3 vs 77.1%, P < 0.01) and parotid gland (13.3 vs 40.9%, P < 0.05) affected. IgG4-CD patients had significantly higher levels of ESR, CRP, IgG, IgG1, IgG3, IgG4, and IgE than IgG4-RD patients. Compared with MCD patients, IgG4-CD patients showed higher incidence of salivary gland and paranasal sinus involvement, higher hemoglobin, eosinophil count, serum IgG4 level and IgG4/IgG ratio, and lower CRP, IL-6 levels, and IgG1/IgG, IgG2/IgG ratio. All patients with IgG4-CD exhibited relatively favorable outcomes. Both IgG4-RD and CD can involve multiple organs. There are a small group of patients who had clinical and pathological characteristics of both CD and IgG4-RD showed better clinical outcome. In the long-term prognosis of these patients, the relationship of CD and IgG4-RD is waiting to be further elucidated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Immunoglobulin G4-related disease (IgG4-RD) is a relatively recently described disease category which affects multiple organs, including the lacrimal gland, salivary gland, parotid gland, lung, pancreas, and retroperitoneum [1, 2]. Diagnosis of IgG4-RD requires a combination of clinical manifestations, serological evidence, and histological performances. Histology is the mainstay of diagnosis, which is characterized by dense immunoglobulin G4 (IgG4)-positive lymphocytes and plasmacyte infiltration, obliterating phlebitis and storiform fibrosis.
Castleman’s disease (CD) is an atypical lymphoproliferative disorder, which can be classified into three types: plasma cell, hyaline vascular, and their mixture. Plasma cell and mixed types frequently have systemic manifestations, which termed multicentric Castleman’s disease (MCD). Patients with MCD often have abnormal laboratory findings, such as hypergammaglobulinemia, elevated C-reactive protein (CRP), interleukin (IL)-6, and vascular endothelial growth factor (VEGF) levels [3,4,5].
It is widely accepted that a diagnosis of IgG4-RD can be made under a premise of ruling out a series of diseases including CD. Actually, sometimes it is really hard to separate the two diseases. In some cases with CD, the histopathological findings, such as IgG4-positive plasma cell infiltrations, are also evident [6,7,8]. Otherwise, IgG4-RD may pathologically exhibit CD-like features. Hence, when the pathological performance meets the diagnosis of both IgG4-RD and CD (IgG4-CD) simultaneously, how can we differentiate the two disease entities? Is it IgG4-related CD or “secondary” IgG4-RD of MCD? Do the clinical course and treatment response differ from those of IgG4-RD or MCD? To our knowledge, no comparative study between IgG4-CD and IgG4-RD, or IgG4-CD and MCD has yet been published. In this study, therefore, we attempted to determine the clinical differences between these three groups of patients using clinical data.
Patients and clinical data
A multidisciplinary collaborative prospective cohort study of IgG4-RD and Mimicry patients has been conducted in Peking Union Medical College Hospital (PUMCH, Beijing, China) since January 2011. This study complied with the Declaration of Helsinki and was approved by the Human and Animal Ethics Review Committees of PUMCH, China. All patients signed written informed consent. Of this 534 patients’ cohort, 15 consecutive patients fulfilled the definition of IgG4-CD. Forty-five definite IgG4-RD patients were enrolled as control. All patients visited the doctors regularly at first and every 3-month intervals. Four- to 6-month follow-up intervals were recommended for patients with long-term stable disease. Up to September 2017, all these patients were followed up over 3 months [30 (3–60 months)].
Patients were diagnosed as definite IgG4-RD according to the 2011 comprehensive diagnostic criteria for IgG4-RD [9]. IgG4-CD were defined histopathologically in patients who fulfilled the criteria of IgG4-RD (IgG4+ plasmacytes > 10/HPF, IgG4/IgG+ plasmacytes > 40%) by pathological features of CD such as atrophic germinal center, mature plasma cell sheet-like proliferation, hyaline vascular lesion, and enlargement of interfollicular area.
Sixteen patients who were diagnosed as plasma cell type MCD without concurrent IgG4-RD were also recruited for comparative analysis. All MCD patients were human immunodeficiency virus (HIV) seronegative, three of them recorded human herpes virus 8 (HHV-8) seronegative, the resting 13 patients had not documented HHV-8 results.
Patients with the following conditions were excluded: infections, malignancies, hypereosinophilic syndromes, and primary vasculitides such as granulomatosis with polyangiitis.
Clinical data and laboratory parameters including complete blood count (CBC), liver and renal function tests, erythrocyte sedimentation rate (ESR), CRP, serum immunoglobulin, IgG subclasses, total IgE, and serum IL-6 (chemiluminescence, SIEMENS) levels were collected. Organ involvements were defined by physical examination or imaging findings including computed tomography (CT), or magnetic resonance imaging (MRI), or positron emission tomography/computed tomography (PET-CT). Using the definitions of the European Academy of Allergy and Clinical Immunology [10], we classified the subjects as either allergic or non-allergic.
Disease relapse was recorded as previously described. In brief, clinical relapse was defined as clinical symptoms recurring or imaging findings getting worsened with or without IgG4 level increasing, and serological relapse was defined as serum IgG4 level getting elevated and IgG4-RD responder index [11] increasing ≥ 1 after improvement with treatment, without recurring clinical symptoms or worsened imaging findings. Overall relapse rate during follow-up embraced both clinical and serological relapses.
Statistical analysis
Continuous variables are described in the standard statistics, including mean and standard deviation. All data were tested for normality and homoscedasticity, then subjected to a Kruskal-Wallis test for data with non-normal distribution or one-way ANOVA for data with normal distribution to determine differences among three groups. All statistical analyses were performed by SPSS version 20.0. A P value < 0.05 was considered statistically significant.
Results
General features of IgG4-CD patients
Fifteen patients (2.8%) fulfilled the diagnosis of IgG4-CD in this study, there were 14 males and 1 female, whose mean age was 47 ± 18 (19–70) years old, and the median disease duration before diagnosis was 12 (1–132) months. Eight patients had allergic history, including allergic dermatitis (3/8), asthma (3/8), allergic rhinitis (1/8), and drug allergy (1/8). Detailed patient profiles are summarized in Table 1. Eleven patients had lymph node (LN) biopsy. The other four patients got pathological specimen at the following sites: submandibular gland plus LN (Nos. 2/3/13) and stomach plus LN (No. 10). Histological examination of HE staining was performed in all tissue samples, as well as immunohistochemical staining with anti-CD3, anti-CD38, anti-CD138, anti-CD20, anti-CD21, anti-IgG, and anti-IgG4 mAb. In all cases, massive lymphocytes and plasma cell infiltration were revealed. IgG4-CD patients were all plasma cell type CD pathologically. The pathological results are shown in Table 2.
Presenting symptoms and organ involvements
The mean age and male/female ratio of control IgG4-RD and MCD patients were 51 ± 14 and 48 ± 15 years, and 32:13 and 9:7, respectively.
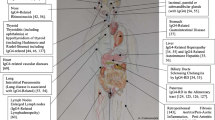
As for the presenting symptoms, lymph node enlargement was most common in IgG4-CD patients, while parotid gland enlargement was prominent in IgG4-RD patients, whereas fever was the most remarkable presenting symptom in MCD patients (P < 0.05) (Fig. 1a).

Presenting symptoms and basement organ involvement of IgG4-CD, IgG4-RD, and MCD patients. a Presenting symptoms of the three groups of patients. b Organ involvement of the three groups of patients. *P < 0.05; **P < 0.01
In terms of organs affected, 100% of IgG4-CD and MCD patients, while 58% of IgG4-RD patients had lymphadenopathy. Submandibular and parotid gland involvements were outstanding features of IgG4-RD patients, compared with the other two groups (Fig. 1b).
Laboratory findings
Summaries of laboratory findings are shown in Table 3. Compared with pure IgG4-RD patients, IgG4-CD patients showed significantly elevated ESR, CRP, IgG, IgG1, IgG3, IgG4, and IgE levels; compared with MCD patients, IgG4-CD patients had higher HGB, eosinophil (Eos), IgG4 levels, and IgG4/IgG ratio, while lower CRP, IL-6 levels, and IgG1/IgG, IgG2/IgG ratios (P < 0.05) (Table 3). Similarly, compared with MCD patients, IgG4-RD patients presented with higher HGB, Eos, IgG4 levels, and IgG4/IgG ratio, and lower ESR, CRP, IgA, IgM, IgG1, IL-6 levels, and IgG1/IgG ratio (P < 0.05) (Table 3).
Thirteen of the 15 patients (86.7%) with IgG4-CD had elevated serum IgG4 levels. Four patients found anemia (adult man < 120 g/L, adult woman < 110 g/L, 6- to 14-year-old child < 120 g/L). Three patients were found hypoalbuminemia (< 35 g/L), and three patients presented with elevated serum IL-6 (3/7, 42.9%).
Clinical outcomes
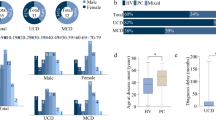
Except for one patient who refused to receive drug therapy was untreated, the remaining patients with IgG4-CD all received glucocorticoid (GC) treatment. Patients with multi-organ involvement or in severe inflammatory condition were treated with both GC and immunosuppressive agents, and those who were with active disease but resistant to above regimen accepted biologics such as rituximab. Six IgG4-CD patients received initial corticosteroid monotherapy, with a median initial dose of equivalent prednisone of 40 mg/days (range 30–60 mg/days). Four patients failed in corticosteroid monotherapy and were added azathioprine, cyclophosphamide or rituximab. Eight IgG4-CD patients received combination therapy of immunosuppressants for the initial treatment, including cyclophosphamide and mycophenolate mofetil. Compared with the initial treatment of IgG4-CD and IgG4-RD patients, no obvious differences were found. Patients were followed up for 33 (4–56) months in IgG4-CD group, and 27 (3–60) months in IgG4-RD group. Follow-up time or intervals did not differ between the two groups. Compared with the poor prognosis of MCD reported in the literature, patients with IgG4-CD exhibited relatively favorable outcomes. The overall relapse rates during the whole follow-up period were 33.3% (including three clinical relapses, two clinical and serological relapses) and 20% (including nine clinical relapses) in IgG4-CD and IgG4-RD patients, respectively (P > 0.05). Higher doses of glucocorticoids or alternative immunosuppressants were used to treat relapsed disease in IgG4-RD group. The fluctuation of serum ESR, CRP, IgE, and IgG4 levels during follow-up are shown in Fig. 2.

Changes of laboratory parameters of IgG4-CD and IgG4-RD patients during follow-up. a–c Changes of ESR, CRP, and IgE levels of the two groups of patients during follow-up. d Gradually descending IgG4 levels in the two groups of patients during follow-up. n, the number of patients in IgG4-CD and IgG4-RD groups shown in different colors
Discussion
To the best of our knowledge, this study is the first relatively big case series to compare the comprehensive clinical characteristics of IgG4-CD and IgG4-RD or MCD, due to the rarity of the three diseases’ condition. We defined patients who synchronously exhibited pathological features of IgG4-RD and CD as IgG4-CD. In China’s largest prospective IgG4-RD and Mimicry study cohort, 15 patients who fulfilled the criteria were found. All of them were human immunodeficiency virus (HIV) negative.
The entanglement of IgG4-RD and CD has a long history. Both the two diseases can involve multiple organs and may have similar clinical and pathological manifestations, such as elevated serum IgG4 and IgG4-positive plasmacyte infiltration in the affected tissue, causing the differential diagnosis of the two diseases in a real dilemma. Lots of efforts had been made at this point to separate the two diseases. It had been reported that orbits, lacrimal glands, salivary glands, pancreas involvement, and complicating allergic disorders were more common in the IgG4-RD patients, whereas the levels of serum IL-6, ESR, CRP, and IgA were significantly higher in the CD patients [12, 13]. Moreover, CD patients had more anemia, hypoalbuminemia, hypocholesterolemia, and thrombocytosis [13, 14]. In the present study, MCD patients presented with higher incidence of fever and LN involvement, and higher levels of ESR, CRP, IgA, IgM, IgG1, IL-6, and IgG1/IgG ratio than IgG4-RD patients, while IgG4-RD patients exhibited elevated incidence of allergic disease history and salivary gland involvement, and higher levels of HGB, Eos, IgG4, and IgG4/IgG ratio than MCD patients. Same as MCD, all IgG4-CD patients had lymph node enlargement (100%); however, IgG4-CD patients presented more frequent involvement of lacrimal gland, submandibular gland, and paranasal sinus, higher HGB, Eos count, serum IgG4 level, and IgG4/IgG ratio, and significantly lower CRP, IL-6 levels, and IgG1/IgG, IgG2/IgG ratio than MCD patients. In addition, there was no difference in allergic disease history between IgG4-CD and IgG4-RD patients. However, IgG4-CD patients had significantly elevated ESR, CRP, IgG, IgG1, IgG3, IgG4, and IgE levels, as compared to IgG4-RD patients. Thirteen patients with IgG4-CD had elevated serum IgG4 levels. Three patients (Nos. 7/11/14) with normal HGB were found elevated serum IL-6, and two of them had slightly decreased serum albumin (Nos. 7/14). Clinically, IgG4-CD patients look more like CD at first glance. But they also had significantly higher incidence of salivary gland involvement, elevated levels of IgG4 and IgE, which are typical characteristics of IgG4-RD. Four out of seven IgG4-CD patients’ tested IL-6 levels were normal. However, it is been universally acknowledged that elevated IL-6 is a characteristic of CD. Therefore, it is hard to phenomenologically classify these IgG4-CD patients as CD or IgG4-RD.
Pathologically, a number of studies had expounded the differences between IgG4-RD and CD. One study in Korean pathologically analyzed 87 CD patients and found that the mean IgG4+/IgG+ plasma cell ratio was 25.1% [15]. Sato et al. [16] reported that the mean IgG4+/IgG+ plasma cell ratio was 57.6% in IgG4-RD patients. They proposed that pathological differentiation of IgG4-RD and CD from lymph node specimens are as follows [16, 17]: (1) the germinal centers of patients with CD were small and regressive, while those of IgG4-RD were normal-hyperplastic; (2) CD showed mature plasma cell sheet-like proliferation in the interfollicular area where IgG4-RD showed proliferation of mature plasma cells with plasmacytoid cells and immunoblasts; (3) IgG4-RD revealed eosinophil infiltration but CD did not. In IgG4-RD patients, the lung lesion was located within the perilymphatic stromal area, and IgG4+/IgG+ cell ratios, eosinophil counts, the extent of active fibrosis, and the number of intravascular lesions were greater. In CD patients, the lung lesion concentrated mainly in the alveolar area besides the perilymphatic stromal area with greater lymphoid follicles [14]. We reviewed the pathological reports of our IgG4-CD patients and found abundant lymphocytes and plasmacyte infiltration in all patients; two patients (Nos. 6/10) showed eosinophil infiltration; four patients (Nos. 2/10/13/15) recorded fibrosis; and two patients (Nos. 10/15) documented obliterating phlebitis.
Our study raises another interesting point regarding the prognosis of IgG4-CD. IgG4-RD usually responds well to glucocorticoids. MCD is somehow a life-threatening disease, and its prognosis is completely different from that of IgG4-RD [16]. The prognosis of IgG4-CD remains unclear, but some studies brought hints that these patients may respond well to glucocorticoids and immunosuppressants. In 2009, Sato et al. [16] examined clinical and pathologic findings of nine patients with systemic IgG4-related lymphadenopathy, three of whom showed CD-like features and one with significantly elevated IL-6. All these three patients were untreated after surgery and remained stable during the follow-up (8–25 months). In 2010, Kojima et al. [18] reported three CD cases of retroperitoneum involvement, with immunohistochemical study demonstrating IgG4(+) plasma cells accounting for more than 50%, two of them with elevated serum IgG4 concentration and normal serum IL-6 levels. One patient responds well to steroids. Two patients were untreated after resection. All three patients were stable during the follow-up period (6–22 months). In 2011, Takenaka et al. [19] described a patient with systemic IgG4-RD whose histopathology mimicked CD was successfully treated with corticosteroids. In 2012, Takeuchi et al. [20] reported a 55-year-old Japanese woman with a 4-year history of sarcoidosis developed erythematous and brown plaques on her back, and she was diagnosed as MCD from a skin biopsy. Laboratory studies revealed anemia, hypoalbuminemia, polyclonal gammaglobulinemia, elevated CRP, and elevated serum IL-6 level. However, serum IgG4 level was remarkably elevated, and the mean IgG4-/IgG-positive cell ratio in her tissue specimens was > 40%, fulfilling the diagnostic criteria of IgG4-RD. Following systemic steroid therapy, the plaques resolved and there was no recurrence observed in 4 months of follow-up. In 2013, Ogoshi et al. [12] reported six CD patients achieved IgG4-RD criteria, four responding well to glucocorticoid monotherapy, one patient with mild disease not receiving any treatment, and one having no response to corticosteroids or repetitive worsening of the symptoms during the tapering of corticosteroids, after being added cyclosporine, following 33 months alive. Very recently, Mochizuki et al. [21] reported a 59-year-old male who was histopathologically diagnosed as CD from his skin lesion with elevated CRP and IL-6 levels, oral prednisolone (PSL) initiated at 0.5 mg/kg relieving his symptoms, but the laboratory data did not improve. After 4 years, he developed lacrimal and salivary gland swelling; histopathological findings in a dacryoadenectomy specimen showed that IgG4+/IgG+ plasma cell ratio was 70%; finally he was diagnosed as IgG4-RD; and rituximab combined PSL treatment were used. Two years after rituximab treatment, the patient’s disease was stable with PSL (0.2 mg/kg). Izumi et al. [22] reported a 50-year-old woman with characteristic features of MCD, including polyclonal hypergammaglobulinemia, multiple enlarged lymph nodes, and pulmonary interstitial infiltration, with elevated serum IgG4 level and a 37.3% IgG4+/IgG+ plasma cell ratio in her biopsy specimen, while without organ involvement of IgG4-RD, corticosteroid treatment resolved the serological and imaging abnormalities. The 14 IgG4-CD patients in present study showed relatively favorable response to glucocorticoid and immunosuppressant therapy, one untreated IgG4-CD patient was stable, during a 33 (4–56)-month follow-up, and this was the same as those reported in the literature.
Our study has some limitations. Although we included patients diagnosed with pathological IgG4-CD in the largest IgG4-RD and Mimicry cohort in China, our results may be affected by institutional bias due to the nature of the single-center study. Secondly, the pathological reports in our hospital were not detailed enough and the biopsy specimens were unaccessible; thus, we failed to do a pathological comparison.
In conclusion, patients with IgG4-CD showed features of both IgG4-RD and MCD. Significantly elevated inflammatory markers in IgG4-CD group patients are similar to those of MCD patients, but those patients also exhibited favorable outcomes as if they were IgG4-RD. It is hard to tell whether it is an overlap, or a subtype of IgG4-RD or CD; therefore, a large cohort study is needed.
References
Umehara H, Okazaki K, Nakamura T, Satoh-Nakamura T, Nakajima A, Kawano M, Mimori T, Chiba T (2017) Current approach to the diagnosis of IgG4-related disease—combination of comprehensive diagnostic and organ-specific criteria. Mod Rheumatol 27(3):381–391. https://doi.org/10.1080/14397595.2017.1290911
Lang D, Zwerina J, Pieringer H (2016) IgG4-related disease: current challenges and future prospects. Ther Clin Risk Manag 12:189–199. https://doi.org/10.2147/TCRM.S99985. eCollection 2016.
Liu AY, Nabel CS, Finkelman BS, Ruth JR, Kurzrock R, van Rhee F, Krymskaya VP, Kelleher D, Rubenstein AH, Fajgenbaum DC (2016) Idiopathic multicentric Castleman’s disease: a systematic literature review. Lancet Haematol 3(4):e163–e175. https://doi.org/10.1016/S2352-3026(16)00006-5
Fajgenbaum DC, Shilling D (2018) Castleman disease pathogenesis. Hematol Oncol Clin North Am 32(1):11–21. https://doi.org/10.1016/j.hoc.2017.09.002.21
Cervantes CE, Correa R (2015) Castleman disease: a rare condition with endocrine manifestations. Cureus 7(11):e380. https://doi.org/10.7759/cureus.380.
Ishida F, Kitano K, Kobayashi H, Saito H, Kiyosawa K (1997) Elevated IgG4 levels in a case with multicentric Castleman’s disease. Br J Haematol 99:981–982
Miwa I, Maruyama Y, Kageoka M, Nagata K, Ohata A, Noda Y, Ikeya K, Matsui T, Koda K, Watanabe F (2008) Retroperitoneal fibrosis and Castleman disease in two patients with high IgG4 levels. Nippon Shokakibyo Gakkai Zasshi 105:1087–1092
Venizelos I, Papathomas TG, Papathanasiou M, Cheva A, Garypidou V, Coupland S (2010) Orbital involvement in Castleman disease. Surv Ophthalmol 55(3):247–255. https://doi.org/10.1016/j.survophthal.2009.09.003
Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T, Matsui S, Yoshino T, Nakamura S, Kawa S, Hamano H, Kamisawa T, Shimosegawa T, Shimatsu A, Nakamura S, Ito T, Notohara K, Sumida T, Tanaka Y, Mimori T, Chiba T, Mishima M, Hibi T, Tsubouchi H, Inui K, Ohara H (2012) Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol 22(1):21–30. https://doi.org/10.1007/s10165-011-0571-z
Johansson SG, Hourihane JO, Bousquet J, Bruijnzeel-Koomen C, Dreborg S, Haahtela T, Kowalski ML, Mygind N, Ring J, van Cauwenberge P, van Hage-Hamsten M, Wuthrich B (2001) A revised nomenclature for allergy. An EAACI position statement from the EAACI nomenclature task force. Allergy 56(9):813–824
Carruthers MN, Stone JH, Deshpande V, Khosroshahi A (2012) Development of an IgG4-RD responder index. Int J Rheumatol 2012:259408. https://doi.org/10.1155/2012/259408
Ogoshi T, Kido T, Yatera K, Oda K, Kawanami T, Ishimoto H, Sakamoto N, Sano A, Yoshii C, Shimajiri S, Mukae H (2013) Assessment of pathologically diagnosed patients with Castleman’s disease associated with diffuse parenchymal lung involvement using the diagnostic criteria for IgG4-related disease. Lung 191(6):575–583. https://doi.org/10.1007/s00408-013-9497-x.
Sasaki T, Akiyama M, Kaneko Y, Mori T, Yasuoka H, Suzuki K, Yamaoka K, Okamoto S, Takeuchi T (2017) Distinct features distinguishing IgG4-related disease from multicentric Castleman’s disease. RMD Open 3(1):e000432. https://doi.org/10.1136/rmdopen-2017-000432
Terasaki Y, Ikushima S, Matsui S, Hebisawa A, Ichimura Y, Izumi S, Ujita M, Arita M, Tomii K, Komase Y, Owan I, Kawamura T, Matsuzawa Y, Murakami M, Ishimoto H, Kimura H, Bando M, Nishimoto N, Kawabata Y, Fukuda Y, Ogura T, Tokyo Diffuse Lung Diseases Study Group (2017) Comparison of clinical and pathological features of lung lesions of systemic IgG4-related disease and idiopathic multicentric Castleman’s disease. Histopathology 70(7):1114–1124. https://doi.org/10.1111/his.13186
Jo JH, Park YS, Jeon YK, Nam SJ, Huh J (2011) Comparison of plasma cell type of Castleman’s disease and IgG4-related sclerosing disease: a histopathological and immunohistochemical study. Pathobiology 78(4):227–232. https://doi.org/10.1159/000327357
Sato Y, Kojima M, Takata K, Morito T, Asaoku H, Takeuchi T, Mizobuchi K, Fujihara M, Kuraoka K, Nakai T, Ichimura K, Tanaka T, Tamura M, Nishikawa Y, Yoshino T (2009) Systemic IgG4-related lymphadenopathy: a clinical and pathologic comparison to multicentric Castleman’s disease. Mod Pathol 22(4):589–599. https://doi.org/10.1038/modpathol.2009.17
Sato Y, Kojima M, Takata K, Morito T, Asaoku H, Takeuchi T, Mizobuchi K, Fujihara M, Kuraoka K, Nakai T, Ichimura K, Tanaka T, Tamura M, Nishikawa Y, Yoshino T (2010) Multicentric Castleman’s disease with abundant IgG4-positive cells: a clinical and pathological analysis of six cases. J Clin Pathol 63(12):1084–1089. https://doi.org/10.1136/jcp.2010.082958
Kojima M, Nakamura N, Motoori T, Shimizu K, Otuski Y, Haratake J, Ogawa A, Igarashi T, Masawa N, Kobayashi H, Nakamura S (2010) Castleman’s disease of the retroperitoneum: with special reference to IgG4-related disorder. J Clin Exp Hematop 50(1):39–44
Takenaka K, Takada K, Kobayashi D, Moriguchi M, Harigai M, Miyasaka N (2011) A case of IgG4-related disease with features of Mikulicz’s disease, and retroperitoneal fibrosis and lymphadenopathy mimicking Castleman’s disease. Mod Rheumatol 21(4):410–414. https://doi.org/10.1007/s10165-010-0410-7.23
Takeuchi M, Sato Y, Takata K, Kobayashi K, Ohno K, Iwaki N, Orita Y, Yoshino T (2012) Cutaneous multicentric Castleman’s disease mimicking IgG4-related disease. Pathol Res Pract 208(12):746–749. https://doi.org/10.1016/j.prp.2012.09.006
Mochizuki H, Kato M, Higuchi T, Koyamada R, Arai S, Okada S, Eto H (2017) Overlap of IgG4-related disease and multicentric Castleman’s disease in a patient with skin lesions. Intern Med 56(9):1095–1099. https://doi.org/10.2169/internalmedicine.56.8013
Izumi Y, Takeshita H, Moriwaki Y, Hisatomi K, Matsuda M, Yamashita N, Kawahara C, Shigemitsu Y, Iwanaga N, Kawakami A, Kurohama H, Niino D, Ito M, Migita K (2017) Multicentric Castleman disease mimicking IgG4-related disease: a case report. Mod Rheumatol 27(1):174–177
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 81373190, 81571587), the Natural Science Foundation of Beijing Municipality (No. 7172178), the Chinese Academy of Medical Sciences Initiative for Innovative Medicine (2017-12M-3-001), and The National Key Research and Development Program of China (No. 2016YFC0901500).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
This study complied with the Declaration of Helsinki and was approved by the Human and Animal Ethics Review Committees of PUMCH, China. All patients signed written informed consent.
Disclosures
None.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhang, X., Zhang, P., Peng, L. et al. Clinical characteristics of a concurrent condition of IgG4-RD and Castleman’s disease. Clin Rheumatol 37, 3387–3395 (2018). https://doi.org/10.1007/s10067-018-4165-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-018-4165-4