
Abstract
Natural Eggshell Membrane (NEM®) is a new novel dietary supplement that contains naturally occurring glycosaminoglycans and proteins essential for maintaining healthy articular cartilage and the surrounding synovium. The randomized, multicenter, double-blind, placebo-controlled Osteoarthritis Pain Treatment Incorporating NEM® clinical study was conducted to evaluate the efficacy and safety of NEM® as a treatment for pain and stiffness associated with osteoarthritis of the knee. Sixty-seven patients were randomly assigned to receive either oral NEM® 500 mg (n = 34) or placebo (n = 33) daily for 8 weeks. The primary endpoint was the change in overall Western Ontario and McMasters Universities (WOMAC) Osteoarthritis Index as well as pain, stiffness, and function WOMAC subscales measured at 10, 30, and 60 days. The clinical assessment was performed on the intent-to-treat population. Supplementation with NEM® produced an absolute rate of response that was statistically significant (up to 26.6%) versus placebo at all time points for both pain and stiffness, but was not significantly improved for function and overall WOMAC scores, although trending toward improvement. Rapid responses were seen for mean pain subscores (15.9% reduction, P = 0.036) and mean stiffness subscores (12.8% reduction, P = 0.024) occurring after only 10 days of supplementation. There were no serious adverse events reported during the study and the treatment was reported to be well tolerated by study participants. Natural Eggshell Membrane (NEM®) is an effective and safe option for the treatment of pain and stiffness associated with knee osteoarthritis. Supplementation with NEM®, 500 mg taken once daily, significantly reduced both joint pain and stiffness compared to placebo at 10, 30, and 60 days. The Clinical Trial Registration number for this study is NCT00750477.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteoarthritis (OA) is the most prevalent form of arthritis and is estimated to affect nearly 27 million adults in the U.S., with one-third of those 65 and older having been diagnosed with OA [1]. As the population ages, this estimate is expected to grow rapidly. Traditional treatments for OA usually involve the use of analgesics (i.e., acetaminophen, tramadol), non-steroidal anti-inflammatory drugs (NSAIDs) (i.e., ibuprofen, diclofenac), or cyclooxygenase-2-specific (COX-2) NSAIDs (i.e., celecoxib) alone or in combination. Steroid and hyaluronic acid injections have also been used with some success. Many of these treatments have shown limited effectiveness in randomized controlled clinical trials (RCTs) [2–5]. To avoid the cardiac risks [6–9] and gastrointestinal issues [10, 11] associated with traditional OA treatments (particularly with long-term use), many patients have turned to complementary and alternative medicines (CAMs) such as dietary supplements.
Glucosamine and chondroitin, alone and in combination, are widely marketed as dietary supplements to treat joint pain due to OA. There have been two major human clinical trials that have investigated the role of these two dietary supplements in the treatment of OA symptoms. The Glucosamine/chondroitin Arthritis Intervention Trial (GAIT), a 1,583-patient, 6-month trial sponsored by the National Institutes of Health (NIH), failed to show significant improvement in the Western Ontario and McMasters Universities (WOMAC) Osteoarthritis Index in the overall patient population for glucosamine, chondroitin, or their combination [12]. The Glucosamine Unum In Die (once-a-day) Efficacy (GUIDE) trial, a 318-patient, 6-month European trial sponsored by industry, showed a small, 5–6% improvement in total WOMAC Index score over placebo for glucosamine sulfate [13]. Because of their limited effectiveness, the search for additional CAMs to treat OA continues.
In the U.S. alone, an estimated 600,000 tons of eggshells are produced annually as a by-product of the poultry industry [14]. Disposal of these eggshells creates an environmental and financial burden and, therefore, alternative uses for these materials would be of obvious benefit. Eggshell membranes are primarily composed of fibrous proteins such as collagen type I [15]. However, eggshell membranes have also been shown to contain glycosaminoglycans (GAGs), such as dermatan sulfate and chondroitin sulfate [16], hexosamines, such as glucosamine, as well as hexoses and fucose [17]. More recently, significant amounts of hyaluronic acid have been detected in eggshell membrane [18]. Other components identified in eggshell membrane include sialic acid [19], desmosine and isodesmosine [20], ovotransferrin [21], lysyl oxidase [22], and lysozyme [23].
The discovery of eggshell membrane as a natural source of combined glucosamine, chondroitin, and hyaluronic acid has prompted the evaluation of this material as a potential treatment for OA. ESM Technologies, LLC (Carthage, MO, USA) has developed methods to efficiently and effectively separate eggshell membrane from eggshells to create a shell-free eggshell membrane. The isolated membrane is then partially hydrolyzed using a proprietary process and dry blended to produce 100% pure Natural Eggshell Membrane (NEM®).
In preliminary open-label human clinical trials totaling 37 subjects with joint and connective tissue disorders, oral supplementation with 500 mg per day of NEM® resulted in an observed decrease in pain in 7–30 days (unpublished report). Therefore, an 8-week randomized, multicenter, double-blind, placebo-controlled supplementation trial was conducted to evaluate the efficacy and safety of NEM® for the relief of the pain and stiffness associated with moderate OA of the knee—the Osteoarthritis Pain Treatment IncorpOrating NEM® (OPTION) trial. The results are presented herein.
Patients and methods
Study design
The OPTION study was conducted according to a randomized, multicenter, double-blind, placebo-controlled design and was conducted in three rheumatology clinics in Missouri (USA) in accordance with the U.S. Food & Drug Administration’s principles of Good Clinical Practice (Title 21, Code of Federal Regulations, Parts 50 & 56 and ICH E6) and the Declaration of Helsinki. The study protocol was approved by an independent regional institutional review board and patients provided their written informed consent to participate. Subjects were required to suspend all current pain relief medications in order to participate in the study. Eligible subjects were then centrally randomized among all sites to receive either NEM® or placebo in the order in which they were enrolled in the study using a permuted-block randomization table consisting of four subjects per block. Patients, clinical investigators, and the study coordinator were all blinded to the treatment through the completion of the study. Treatment consisted once daily orally of either NEM® (Membrell, LLC, USA) or placebo (excipients) provided in 500 mg vegetarian capsules that were stored in closed containers at ambient temperature. Clinic visits were scheduled for subjects at 10, 30, and 60 days following the onset of treatment. Treatment compliance was checked at clinic visits by patient interview and by counting the number of unused doses of the study medications. Acetaminophen was allowed for pain relief rescue, if necessary. Subjects recorded the time and amount of acetaminophen taken in patient diaries.
Patients
All subjects 18 years of age or older with known symptomatic osteoarthritis of the knee were considered for enrollment in the study. In order to be eligible, patients must have been diagnosed with functional grades I–III of osteoarthritis according to the modified criteria of the American College of Rheumatology [24]. Subjects must also have had persistent knee pain associated with osteoarthritis with a baseline score of at least 30 mm on the Patient’s Assessment of Arthritis Pain—Visual Analog Scale. Subjects were required to suspend all current pain relief medications. Subjects that were currently taking analgesic medications were eligible to participate in the study following a 14-day washout period for NSAIDs, a 7-day washout for narcotics, and a 90-day washout for injected steroids. Subjects currently taking glucosamine, chondroitin sulfate, or MSM were only eligible after a 3-month washout period. Patients were excluded if they were currently receiving remission-inducing drugs such as methotrexate or immunosuppressive medications or had received them within the past 3 months. They were also excluded if they had a confounding inflammatory disease or condition (rheumatoid arthritis, gout, pseudo gout, Paget’s disease, chronic pain syndrome, etc.) that would interfere with assessment of pain associated with the index knee. Other exclusionary criteria were: body weight 113.5 kg (250 lbs) or greater, a known allergy to eggs or egg products, or pregnant or breastfeeding women. Subjects previously enrolled in a study to evaluate pain relief within the past 6 months or currently involved in any other research study involving an investigational product (drug, device, or biologic) or a new application of an approved product, within 30 days of screening, were also excluded from participating in the trial.
Treatment response
The primary endpoint of the study was measurement of the effectiveness of NEM® in relieving pain, stiffness, and discomfort associated with moderate OA of the knee and to compare its effectiveness to placebo. The primary treatment response endpoints were the 10-, 30-, and 60-day clinic assessments utilizing the Western Ontario and McMasters Universities (WOMAC) Osteoarthritis Index—Visual Analog Scale (100 mm) version (VA 3.1) [25]. This version of the WOMAC questionnaire consists of five questions addressing the severity of joint pain, two questions addressing joint stiffness, and 17 questions addressing limitations in performing physical activities (function). Endpoints were compared to pretreatment assessments and to placebo controls. In 2004, the Outcome Measures in Rheumatology Clinical Trials (OMERACT) and the Osteoarthritis Research Society International (OARSI) published criteria for a response to treatment for osteoarthritis [26]. A treatment response was classified as an improvement in pain or function of at least 50% and a decrease of at least 20 mm on the visual-analog scale for pain or function (WOMAC subscales). Alternatively, the occurrence of two of the following criteria also was acceptable as a treatment response: a decrease in pain of at least 20% and at least 10 mm on the visual-analog scale; an improvement in function of at least 20% and a decrease of at least 10 mm on the visual-analog scale; and an increase in the patient’s global assessment score by at least 20% and at least 10 mm on the visual-analog scale. Patient’s global assessment scores were not collected using a visual-analog scale; however, we only review the OMERACT–OARSI response rate with respect to the primary criterion. Patient’s and Physician’s Global Assessments of Arthritis were also collected utilizing a 0–5-point Likert scale.
Adverse events
Secondary objectives of the study were to evaluate tolerability and any adverse reactions associated with supplementation with NEM®. The subjects’ self-assessment diaries were reviewed and any discomfort or other adverse events were recorded and reported in accordance with applicable FDA regulations. Adverse events and serious adverse events were assessed by the clinical investigator at each study visit and followed until resolution, as necessary. Serious adverse events were required to be reported to the clinical monitor immediately using MedWatch OMB No. 0910-0291.
Statistical analysis
An absolute increase in the mean response rate of 35% (treatment rate versus placebo rate) was considered a clinically meaningful treatment effect. It was estimated that a sample size of 75 patients would need to be enrolled to provide the study with a statistical power of 85% to detect a clinically meaningful difference between the treatment group and the placebo group, assuming a rate of response of 40% in the treatment group, a rate of response of 5% in the placebo group, and a withdrawal rate of 20%. Pairwise comparisons of the treatment group with the placebo group were made with a two-sided independent group t test at baseline to validate randomization. Within-group comparisons, using a two-sided independent group t test, were also made between testing sites to rule out any site bias. In both cases, statistical significance was accepted at an α value of <0.05. Post-baseline statistical analyses were done utilizing repeated measures univariate analysis of variance (RM-ANOVA) on pooled population data. Statistical significance was accepted at an α value of <0.05 for between-group interactions. Analysis of the primary outcome measure (the difference between groups and the change from baseline in overall WOMAC composite score as well as pain, stiffness, and function subscores) was conducted in the intent-to-treat (ITT) population (i.e., including all randomized patients with at least one efficacy assessment after randomization). The last observation carried forward (LOCF) approach was used for patients who made at least one follow-up visit but who did not complete the study (lost to follow-up). A per-protocol completer analysis was also performed. SYSTAT software (version 12) was used for all statistical analyses [27].
Results
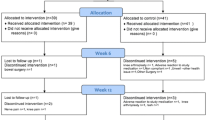
Patient recruitment began in December 2004 at three clinical sites in Missouri and the final follow-up was conducted in January 2006. A total of 67 subjects were enrolled in the trial and underwent randomization (see Fig. 1). Of these subjects, 61.1% were from site 1, 29.9% from site 2, and 9.0% from site 3. In terms of OA functional grades, 20.9% were grade I, 28.4% were grade II, 20.9% were grade III, and 29.9% were unassigned. Seven subjects did not complete baseline evaluations, resulting in a total of 60 subjects in the intent-to-treat (ITT) population. Thirty-one subjects (51.6%) were randomized to the placebo group and 29 subjects (48.3%) were randomized to the NEM® treatment group. Thirty-one percent (31%) of the ITT subjects assigned to NEM® did not complete the 2-month study per the protocol, compared with 42% of the ITT subjects assigned to placebo. Of the 60 subjects in the ITT population, six subjects assigned to placebo and two subjects assigned to NEM® either violated the protocol or did not begin treatment and, therefore, were not available for further analysis. Those patients lost to follow-up before the first evaluation time point in both the placebo (four patients) and treatment (three patients) groups had symptomatically mild OA (mean WOMAC 39.7 and 45.6, respectively). Those patients lost to follow-up (primarily withdrawals) in the remainder of the study in both the placebo (three patients) and the treatment (four patients) groups had symptomatically more severe OA (mean WOMAC 76.6 and 63.7, respectively) compared to those patients that completed the study (mean WOMAC at baseline of 52.6 and 45.3, respectively). Five patients in the placebo group and four patients in the treatment group officially withdrew from the study due to lack of efficacy. There were no obvious differences in the reason for withdrawal between the study groups. The overall drop-out rate of 43% (from enrollment) was considerably higher than the estimated rate of 20%, although >70% of the subjects that began treatment (n = 54) completed the study (excluding non-compliance). This is possibly related to the small sample population or could also be due to the relatively stringent pain management requirements of the study protocol. Compliance with the study treatment regimen was good in both treatment groups. In those subjects that completed the study, the rate of compliance was >97% (as judged by capsule count at clinic visits).

Enrollment, randomization, and completion flow diagram
Patient data was initially evaluated to ensure randomization within each site. Additionally, patient data was evaluated between sites to exclude site bias. Due to the lack of a characteristic placebo effect in the overall study population, blinding was further scrutinized within the placebo group. When evaluating the placebo group by site and month enrolled, the placebo effect that was observable was evenly distributed over time and between sites. That is, there was a relevant placebo effect in six of the eight months of enrollment across all sites. As there were no observable systematic abnormalities in any of these evaluations, the data were pooled for all subsequent analyses. A clinical comparison of valid (excluding non-compliance) subjects was carried out to obtain mean baseline values (see Table 1). In all cases, the treatment group values were slightly lower than those of the control group, but were not statistically different. Analysis of the primary outcome measure revealed that supplementation with NEM® produced an absolute rate of response that was significantly better (ranging from 10.3% to 26.6% improvement) than placebo at all time points for both pain and stiffness, but fell short of significance for function and overall WOMAC, despite improving by 8.8% to 15.5% (see Table 2). There were rapid responses seen for mean pain subscores (15.9% reduction, P = 0.036) and mean stiffness subscores (12.8% reduction, P = 0.024) occurring after only 10 days of supplementation. At 60 days, pain response was maintained (15.4%, P = 0.038), while stiffness had improved further to 26.6% reduction (P = 0.005). Mean function subscores showed a 15.5% (P = 0.084) absolute improvement versus placebo at 10 days, which fell slightly to 13.5% (P = 0.076) by day 60. Overall mean WOMAC scores resulted in a 15.2% (P = 0.059) absolute improvement versus placebo at 10 days, which was maintained at 60 days (15.1%, P = 0.052). Thirty-two percent (32%) of patients in the treatment group demonstrated a primary OMERACT–OARSI response versus 12% of patients in the placebo group by the end of the follow-up period (60 days, P = 0.023).
The study population was too small to stratify the patients according to covariates, such as baseline pain level or grade (I–III), to obtain statistically relevant data. There was little difference between response rates for patients with more severe OA (grades II/III) compared with patients with less severe OA (grade I). About one-third of the patients in both groups had at least a 40% reduction in pain at 60 days.
Overall, the use of rescue pain medication was low throughout the study, occurring once in every 6–8 days. There were no significant differences among the groups for rescue medication use. Approximately 17% of subjects took acetaminophen significantly more often than average (once every 3 days or less). After reviewing the patient diaries, it was noted that many of the instances of acetaminophen use were not for rescue purposes, but were actually for headaches, backaches, and other issues unrelated to the treatment knee. There were three adverse events reported during the study and none of them were judged by clinical investigators to be associated with treatment. There were no serious adverse events reported during the study. Of particular note is that there were no allergy-associated adverse events during the study, although those with known egg allergies were excluded from participating during screening. In general, the treatment was reported to be extremely well tolerated by study participants.
Discussion
Osteoarthritis is extremely prevalent and results in significant costs, both financial and quality of life, for those that suffer from the debilitating disease. The OPTION trial was designed to evaluate the efficacy and safety of Natural Eggshell Membrane as a treatment option for osteoarthritis. Our preliminary study indeed proved NEM® both effective and safe for treating pain and stiffness associated with OA of the knee. NEM® has the added benefit of avoiding the concerning side effects associated with long-term use of other OA treatments such as NSAIDs.
Patients experienced a relatively rapid (10 days) response for all WOMAC scores with a mean response of approximately 15% (12.8% to 15.9%). By the end of the follow-up period (60 days), the mean response remained approximately 15% (13.5% to 15.4%) for all WOMAC scores except stiffness which was 26.6%. While this is superior to the response shown for glucosamine and chondroitin in previous clinical investigations [12, 13], it failed to reach the expected 35% response rate employed in the clinical design. Despite this shortcoming, the results were shown to be statistically significant. The safety profile for NEM® is also of significance as there are no known side effects, excluding the obvious egg allergy concern. This is of obvious importance in a condition that requires long-term treatment. Significant and sometimes serious side effects associated with other OA treatments frequently limits treatment options.
The measure of subjective symptoms (i.e., pain, stiffness, etc.) of arthritis and the wide variation in individual patient’s perception of these symptoms results in complex relationships that can be difficult to elucidate from the reporting of mean treatment effects in clinical trials and may fail to adequately describe the potential benefits to the individual patient [28–31].
Number Needed to Treat (NNT) is a form of responder analysis and is a widely accepted and statistically valid measure of treatment effect [32]. NNTs of 5 or below are generally accepted as equating to an effective treatment for pain-related conditions [30]. In order to perform an NNT evaluation of the OPTION data, a treatment response rate table was prepared for the treatment and placebo groups at all time points for the pain (see Table 3) and stiffness (not shown) WOMAC subscales. It becomes evident that there are response rates that are quite likely to be clinically relevant (i.e., ≥30% reduction from baseline). For example, approximately one-third (33%) of study subjects experienced greater than 30% reduction in pain at 10 days, with a similar number of subjects (32%) having experienced greater than 50% reduction in pain at 60 days. In both instances, this rate was more than two times (∼2.5×) that of the placebo group. Approximately one-quarter (25%) of study subjects experienced greater than 50% reduction in stiffness at 10 days, with the number of patients increasing to more than one-half (53%) having experienced this level of improvement at 60 days. The 10-day result was more than two times (∼2.5×) that of the placebo group and the 60-day result was nearly five times (∼4.8×) that of placebo.
These various responder rates were then converted to NNT values which include 95% confidence intervals (95% CI) according to the method described by Wen et al. [33]. NNT values were determined for each level of improvement (as shown in Table 3) for both pain and stiffness. At 10, 30, and 60 days, NNTs for at least 50% reduction in pain were 28.0 (95% CI, 26.2 to 29.8), 5.6 (3.9 to 7.4), and 5.0 (3.1 to 6.9), respectively. In clinical practice, one out of every five patients should experience at least a 50% reduction in pain within 30–60 days. By comparison, we determined an NNT of 23.8 (95% CI, 15.2 to 32.4) from the GAIT data for a 50% reduction in WOMAC pain scores for the overall study population [12]. A similar 50% reduction in rheumatoid arthritis pain was reported as 4 in a review of three clinical trials for adalimumab, etenercept, and double-dose infliximab [34].
NNT values were also determined for 50% reduction in stiffness at each time point. We obtained NNTs of 6.5 (95% CI, 4.6 to 8.4), 7.9 (6.1 to 9.7), and 2.4 (0.5 to 4.3) at 10, 30, and 60 days, respectively. This demonstrates that there is a clinically relevant reduction in stiffness at all time points during the study. This is particularly true at 60 days where nearly one out of every two patients would experience a 50% reduction in stiffness.
With one-third of those 65 and older having been diagnosed with osteoarthritis [1], and that number expected to grow immensely as the overall US population ages, it is important for patients to have treatment options that are both effective and safe. The reporting of the results from the OPTION trial provides this needed treatment option.
The trial suffered from a number of issues. The limited initial enrollment (67 subjects), the relatively high drop-out rate (43%), and the smaller mean treatment effect than anticipated (15% versus 35%) could have compromised the statistical significance of the trial results. In addition to these inherent limitations, combined they also prevented post hoc analysis of subgroups of patients, say by severity of disease. Less stringent requirements for concomitant pain treatment or a more liberal rescue pain policy may have reduced the drop-out rate considerably. The inclusion of a comparative treatment agent may have provided additional information, but would have required a significantly larger study population. A larger follow-up study with some modifications may allow us to better determine which patients are most helped by NEM® supplementation.
References
Lawrence RC, Felson DT, Helmick CG et al (2008) Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum 58(1):26–35
Case JP, Baliunas AJ, Block JA (2003) Lack of efficacy of acetaminophen in treating symptomatic knee osteoarthritis. Arch Intern Med 163:169–178
Towheed TE, Maxwell L, Judd MG, Catton M, Hochberg MC, Wells G (2006) Acetaminophen for osteoarthritis. Cochrane Database of Systematic Reviews, No. 2
Geba GP, Weaver AL, Polis AB, Dixon ME, Schnitzer TJ (2002) Efficacy of rofecoxib, celecoxib, and acetaminophen in osteoarthritis of the knee. J Am Med Assoc 287(1):64–71
Altman RD (1999) Ibuprofen, acetaminophen and placebo in osteoarthritis of the knee: a six-day double-blind study [abstract]. Arthritis Rheum 42:S403
Bresalier RS, Sandler RS, Quan H et al (2005) Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med 352:1092–1102
Solomon SD, McMurray JJ, Pfeffer MA et al (2005) Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med 352:1071–1080
Nussmeier NA, Whelton AA, Brown MT et al (2005) Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N Engl J Med 352:1081–1091
Singh G, Wu O, Langhorne P, Madhok R (2006) Risk of acute myocardial infarction with nonselective non-steroidal anti-inflammatory drugs: a meta-analysis. Arthritis Res Ther 8(5):153–162
Deeks JD, Smith LA, Bradley MD (2002) Efficacy, tolerability, and upper gastrointestinal safety of celecoxib for treatment of osteoarthritis and rheumatoid arthritis: systematic review of randomised controlled trials. Br Med J 325:619–627
Laine L (1996) Nonsteroidal anti-inflammatory drug gastropathy. Gastrointest Endosc Clin N Am 6(3):489–504
Clegg DO, Reda DJ, Harris CL et al (2006) Glucosamine, chondroitin sulfate, and the two in combination for painful knee osteoarthritis. N Engl J Med 354(8):795–808
Herrero-Beaumont G, Ivorra JAR, Trabado MC et al (2007) Glucosamine sulfate in the treatment of knee osteoarthritis symptoms. Arthritis Rheum 56(2):555–567
U.N. Food & Agriculture Organization: FAO Statistical Yearbook—United States of America (2004)
Wong M, Hendrix MJC, von der Mark K et al (1984) Collagen in the egg shell membranes of the hen. Devel Biol 104(1):28–36
Baker JR, Balch DA (1962) A study of the organic material of hen's-egg shell. Biochem J 82:352–361
Picard J, Paul-Gardais A, Vedel M (1973) Sulfated glycoproteins from egg shell membranes and hen oviduct. Isolation and characterization of sulfated glycopeptides. Biochimica et Biophysica Acta 320:427–441
Long FD, Adams RG, and DeVore DP (September 20, 2005) Preparation of hyaluronic acid from eggshell membrane. USA Patent #6946551
Nakano T, Ikawa NI, Ozimek L (2003) Chemical composition of chicken eggshell and shell membranes. Poult Sci 82:510–514
Starcher BC, King GS (1980) The presence of desmosine and isodesmosine in eggshell membrane protein. Connect Tissue Res 8:53–55
Gautron J, Hincke MT, Panheleux M et al (2001) Ovotransferrin is a matrix protein of the hen eggshell membranes and basal calcified layer. Conn Tissue Res 42:255–267
Akagawa M, Wako Y, Suyama K (1999) The presence of desmosine and isodesmosine in eggshell membrane protein. Biochim Biophys Acta 14:151–160
Hincke MT, Gautron J, Panheleux M et al (2000) Identification and localization of lysozyme as a component of eggshell membranes and eggshell matrix. Matrix Biol 19:443–453
Hochberg MC, Chang RW, Dwosh I, Lindsey S, Pincus T, Wolfe F (1992) The American College of Rheumatology 1991 revised criteria for the classification of global functional status in rheumatoid arthritis. Arthritis Rheum 35:498–502
Bellamy N, Buchanan WW, Goldsmith CH, Campbell J, Stitt LW (1988) Validation study of WOMAC: a health status instrument for measuring clinically important patient relevant outcomes to antirheumatic drug therapy in patients with osteoarthritis of the hip or knee. J Rheumatol 15:1833–1840
Pham T, van der Heijde D, Altman RD et al (2004) OMERACT-OARSI initiative: Osteoarthritis Research Society International set of responder criteria for osteoarthritis clinical trials revisited. Osteoarthritis Cartilage 12:389–399
Systat Software, Inc. , San Jose, CA, USA: www.systat.com
Tubach F, Ravaud P, Giraudeau B (2005) Managing osteoarthritis of the knee: conclusions about use of NSAIDs are misleading. Br Med J 330:672
McQuay H, Moore A (2007) Utility of clinical trial results for clinical practice. Eur J Pain 11:123–124
Moore RA, Moore OA, Derry S, McQuay HJ (2008) Numbers needed to treat calculated from responder rates give a better indication of efficacy in osteoarthritis trials than mean pain scores. Arthritis Res Ther 10(2):5
Lötsch J, Geisslinger G (2006) Current evidence for a genetic modulation of the response to analgesics. Pain 121:1–5
Saver JL (2004) Number needed to treat estimates incorporating effects over the entire range of clinical outcomes. Arch Neurol 61:1066–1071
Wen L, Badgett R, Cornell J (2005) Number needed to treat: a descriptor for weighing therapeutic options. Am J Health Syst Pharm 62(19):2031–2036
Kristensen LE, Christensen R, Bliddal H, Geborek P, Danneskiold-Samsøe B, Saxne T (2007) The number needed to treat for adalimumab, etanercept, and infliximab based on ACR50 response in three randomized controlled trials on established rheumatoid arthritis: a systematic literature review. Scand J Rheumatol 36:411–417
Funding
This study was supported by ESM Technologies, LLC.
Disclosures
KJR is currently employed by the sponsor of the study. AW and RWJ have no competing interests. DPD and BWR have served as paid consultants for the sponsor of the study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Key Messages
Natural Eggshell Membrane (NEM®), 500 mg taken once daily, significantly reduced both joint pain and stiffness compared to placebo, both rapidly (10 days) and continuously (60 days).
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Ruff, K.J., Winkler, A., Jackson, R.W. et al. Eggshell membrane in the treatment of pain and stiffness from osteoarthritis of the knee: a randomized, multicenter, double-blind, placebo-controlled clinical study. Clin Rheumatol 28, 907–914 (2009). https://doi.org/10.1007/s10067-009-1173-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-009-1173-4