
Abstract
Biallelic (autosomal recessive) pathogenic variants in ATP13A2 cause a form of juvenile-onset parkinsonism, termed Kufor-Rakeb syndrome. In addition to motor symptoms, a variety of other neurological and psychiatric symptoms may occur in affected individuals, including supranuclear gaze palsy and cognitive decline. Although psychotic symptoms are often reported, response to antipsychotic therapy is not well described in previous case reports/series. As such, we describe treatment response in an individual with Kufor-Rakeb syndrome-associated psychosis. His disease was caused by a homozygous novel loss-of-function ATP13A2 variant (NM_022089.4, c.1970_1975del) that was characterized in this study. Our patient exhibited a good response to quetiapine monotherapy, which he has so far tolerated well. We also reviewed the literature and summarized all previous descriptions of antipsychotic treatment response. Although its use has infrequently been described in Kufor-Rakeb syndrome, quetiapine is commonly used in other degenerative parkinsonian disorders, given its lower propensity to cause extrapyramidal symptoms. As such, quetiapine should be considered in the treatment of Kufor-Rakeb syndrome-associated psychosis when antipsychotic therapy is deemed necessary.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Biallelic pathogenic variants in ATP13A2, also known as PARK9, are associated with an autosomal recessive form of juvenile-onset parkinsonism, termed Kufor-Rakeb syndrome (KRS). Additional clinical features include facial-faucial-finger myoclonus, supranuclear gaze palsy, oculogyric dystonic spasms, and dementia, in addition to various neuropsychiatric symptoms, including psychosis [1, 2]. Some affected individuals are considered to have a form of neurodegeneration with brain iron accumulation or neuronal ceroid lipofuscinosis [1, 2], and spastic paraplegia has also been described in relation to recessive ATP13A2 variants [3].
ATP13A2 encodes for the ATPase 13A2 (ATP13A2). ATP13A2 belongs to the superfamily of P-type ATPase transporters, which are alternatingly auto-phosphorylated and dephosphorylated on a conserved aspartic acid during their catalytic cycle [4]. ATP13A2 has recently been described as a polyamine transporter with highest affinity for the polyamines spermine and spermidine. At the cellular level, ATP13A2 is localized in late endolysosomes, where it exports the endocytosed polyamines spermine and spermidine from the lumen to the cytosol [5]. Polyamines are neuroprotective agents and are involved in a plethora of pathways, ranging from the regulation of transcription and translation to autophagy and anti-oxidant responses [6]. As ion channel modulators, polyamines have been implicated in several mental health problems/disorders, including schizophrenia, mood disorders, anxiety, and suicidal behaviour [7]. ATP13A2 loss-of-function causes a disturbed intracellular polyamine distribution with (i) polyamine accumulation in late endolysosomes, resulting in rupture of these organelles and cathepsin B-mediated cell death [5], and (ii) a deficiency of cytosolic polyamines, leading to more mitochondrial derived reactive oxygen species causing oxidative stress [8]. Interestingly, all KRS variants characterized up until now show a (nearly) complete loss of ATP13A2 function as a consequence of protein mislocalization, instability, hampered autophosphorylation, dephosphorylation, and/or ATPase activity, or a combination thereof [3, 5, 9,10,11,12,13].
Although psychotic symptoms may occur in individuals with KRS (even in the absence of dopaminergic therapy), response to antipsychotic treatment has infrequently been described in the literature. As particular recommendations regarding the treatment of psychosis in this population remain scarce, we report a proband with KRS-associated psychosis caused by a homozygous novel loss-of-function ATP13A2 variant, who ultimately responded well to quetiapine monotherapy. We also performed a review of the literature with respect to treatment response in individuals with psychosis.
Case report
We report a 22 year old male with a mild intellectual disability (FSIQ = 73 in 2016) who presented to clinical attention after abruptly developing psychotic symptoms in 2019 at the age of 17. He was born around 36 weeks gestation via cesarean section due to either oligohydramnios or breech position (there have been conflicting reports in this respect). Although there were no gross motor delays he was described as being clumsier than his peers and later struggled with handwriting. He was delayed in both expressive and receptive speech and had articulation difficulties. Although he did not undergo a formal psychoeducational assessment until his teenage years (in 2016), he reportedly always struggled academically. Starting at around the time of his psychiatric admission in 2019 (but possibly earlier), his cognitive abilities began to progressively deteriorate. He nonetheless completed grade 12 thereafter, following his admission to hospital. Frank motor symptoms had not been identified or endorsed prior to the initiation of antipsychotic medication (see below for details). His medical history is otherwise only remarkable for intermittent low grade microcytic anemia and bilateral hydroceles (for which he underwent a bilateral hydrocelectomy in 2022).
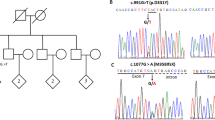
In terms of family history, he has a paternal first cousin who is intellectually disabled and non-ambulatory; however, her presentation is thought to be non-progressive. He has two additional paternal first cousins (who are siblings) who are autistic and/or intellectually disabled, and non verbal. There is otherwise no known history of parkinsonism, psychosis, or any other neuropsychiatric syndromes in any family members. A pedigree is provided in Fig. 1.

Pedigree
Apart from vaguely described anxiety, of a generalized and social nature, as well as possible attention-deficit/hyperactivity disorder, he was generally psychiatrically well prior to his index admission to hospital in 2019 (at age 17), which occurred immediately following a traumatic event. Although his symptoms were initially queried to represent a severe trauma reaction, the presence of frank psychotic symptoms eventually became evident, including disorganization of thought and behaviour, as well as persecutory, referential, and hyperreligious delusions that occurred in association with thematically congruent auditory hallucinations. For example, he became concerned that his father was “the devil”, and reported hearing “Satan” and “Jesus” speaking to him. In this context, he was also observed responding to internal stimuli. While his psychotic symptoms initially resolved with the initiation of low dose risperidone, he experienced remarkably severe extrapyramidal symptoms (EPS). As such, he was transitioned to olanzapine in the months following his discharge, which was comparatively less problematic from a motor side effect standpoint, but nonetheless continued to cause atypically severe EPS on doses as low as 2.5 mg qhs.
While a general medical workup was unrevealing, including a brain MRI which did not show obvious evidence of neuronal brain iron accumulation, his overall neuropsychiatric and developmental picture combined with a history of parental consanguinity, prompted a medical genetics service consult shortly before his discharge from hospital. While initial testing with chromosomal microarray was negative, clinical exome sequencing (Blueprint Genetics 2021) revealed a homozygous variant in ATP13A2 (NM_022089.4, c.1970_1975del) which is predicted to cause an in-frame deletion of two amino acids, p.(Pro657_Glu658del). This variant is rare and absent from the gnomAD v4 database (accessed Nov 28, 2023). Although this in-frame deletion was initially formally classified as a variant of uncertain significance, it was clinically considered to be likely pathogenic given that his overall presentation was highly suggestive of KRS. Additionally, biochemical analysis demonstrating that the variant causes loss-of-function was performed (see below for details), confirming pathogenicity.
While his psychotic symptoms and comorbid anxiety were initially reasonably well controlled with low dose olanzapine following his discharge from hospital, he exhibited worsening dysarthria, dysphagia, bradykinesia, and balance difficulties over the subsequent years (between 2020 and 2022). He was assessed in a specialized movement disorders clinic in 2022 at age 20 and was found to exhibit new-onset marked difficulties with vertical eye movements in the upward plane with hypometric saccades in the horizontal plane. At the time of the assessment there was no blepharospasm and no orofacial dystonia. Marked hypomimia and hypophonia were noted. Moderate rigidity was noted axially as well as in both upper extremities with marked (slightly asymmetric) bradykinesia in all four limbs (more prominent in the left upper extremity and right lower extremity). He had no difficulty standing up from a seated position. Gait examination revealed markedly reduced stride length and arm swing, particularly in the left upper extremity. The pull test was positive. Deep tendon reflexes were brisk in the upper extremities at 3 + with no Hoffmann’s sign, and 2 + at the knees. No spasticity was appreciated. No tremor or any other abnormal movements were observed. On more recent examinations (beginning in early 2023), facial-faucial-finger mini-myoclonus has also been observed.
When attempts were made to lower his dose of olanzapine to 1.25 mg qhs, his motor symptoms predictably improved but he became more paranoid and anxious. As such, he was eventually cross-titrated from olanzapine to quetiapine in late 2022 at the age of 21. Although he experienced a reemergence of symptoms at certain points during the transition to quetiapine, he eventually again achieved remission of his psychotic symptoms on quetiapine monotherapy (initially on quetiapine XR 200 mg qhs in addition to quetiapine IR 100 mg qhs, before his dose of the XR formulation was increased to 300 mg due to a recurrence of occasional mild stress-induced paranoia). A combination of the extended release and immediate release formulations was chosen, as he experienced more frequent breakthrough symptoms in a diurnal pattern (specifically, in the evenings prior to his next dose) on the immediate release formulation alone. Additionally, as expected his motor symptoms noticeably improved with the change to quetiapine, despite some degree of ongoing dysarthria, rigidity, and bradykinesia, as well as continued reduced stride length and arm swing while walking.
Methods
Biochemical analysis of the ATP13A2 variant
Cell culture - Human neuroblastoma SH-SY5Y (RRID: CVCL_0019) cell lines either non-transduced (nts) or stably overexpressing wild-type ATP13A2 (Addgene plasmid #171485, RRID: Addgene_171485 or Addgene plasmid #213697, RRID: Addgene_213697), a catalytically dead variant (D508N) (Addgene plasmid #171820, RRID: Addgene_171820), or the Pro652_Glu653del ATP13A2 variant (Addgene plasmid #213700, RRID: Addgene_213700) (the nomenclature is based on the sequence of ATP13A2 splice variant 2, which is 5 amino acids shorter at the N-terminal region and has been historically used for biochemical analysis [5]) were generated via lentiviral transduction as described previously [14]. A detailed protocol can be found at https://doi.org/10.17504/protocols.io.bw57pg9n. Cells were maintained at 37 °C in the presence of 5% CO2 and incubated in high-glucose Dulbecco’s modified Eagle medium supplemented with 1% penicillin/streptomycin (Sigma), 15% fetal calf serum (heat inactivated) (Sigma), 1% non-essential amino acids (Sigma), 1% sodium pyruvate (Gibco), and selection antibiotic (160 µg/mL hygromycin or 2 µg/mL puromycin (Invivogen)). The treatments were performed in the same medium, with the exclusion of selection antibiotic.
Immunofluorescence - Cells were seeded at 75 000 cells/well in a 12-well plate with coverslips and allowed to attach and grow for 48 h. Thereafter, cells were washed with ice-cold PBS and fixed with 4% paraformaldehyde (Thermo Fisher Scientific) (30 min, 37 °C). Cells were washed twice more with ice-cold PBS before permeabilization and blocking with a mixture of 5% BSA (Roth) and 0.5% saponin (Sigma) (referred to as blocking buffer) for 1 h. Next, cells were incubated with primary antibody (anti-ATP13A2, A3361, Sigma, RRID: AB_10597403; anti-CD63, 11-343-C100, ExBio, RRID: AB_10733918) (diluted 1/200 in blocking buffer) for 2 h and then subjected to secondary antibody (Alexa-488 goat anti-rabbit IgG, R37116, Invitrogen, RRID: AB_2556544; Alexa-594 goat anti-mouse IgG, A11005, Invitrogen, RRID: AB_2534073) (diluted 1/1000 in blocking buffer) for 30 min on a shaker. Subsequently, cells were immersed in 200 ng/ml DAPI (D9542, Sigma) for 15 min. Samples were thoroughly washed with PBS in between the different steps. Finally, the samples were mounted and images were acquired using a LSM880 microscope (Zeiss) with a 63x objective and Airyscan detector. A detailed protocol can be found at: https://doi.org/10.17504/protocols.io.bp2l6xy3klqe/v1.
SDS-page and immunoblotting – 70% confluent cells were harvested and subsequently lysed with radio-immunoprecipitation assay buffer (Thermo Fisher Scientific) supplemented with protease inhibitors (Sigma). 10 µg of protein (concentration determined with a bicinchoninic acid protein assay) was then loaded on a NuPage 4–12% Bis-Tris gel (Thermo Fisher Scientific, Bio-Rad) and subjected to an electrophoresis run at 130 V in MES running buffer (Thermo Fisher Scientific). The proteins were subsequently transferred to a PVDF membrane in NuPage transfer buffer (Thermo Fisher Scientific) supplemented with 10% v/v methanol (Roth). Immunoblots were blocked by incubation in 5% w/v milk powder (1 h, room temperature). Next, immunoblots were probed with primary antibodies (anti-GAPDH, G8795, Sigma, RRID: AB_1078991; anti-ATP13A2, A3361, Sigma, RRID: AB_10597403) (diluted 1/5000 and 1/1000 in 1% w/v BSA, respectively) and incubated O/N (4 °C). After thorough washing, immunoblots were subjected to secondary antibodies (HRP-linked anti-mouse IgG, 7076 S, Cell Signaling, RRID: AB_330924; HRP-linked anti-rabbit IgG, 7074 S, Cell Signaling, RRID: AB_2099233) (diluted 1/2000 in 1% w/v milk powder) (1 h, room temperature). All dilutions and wash steps were performed with TBS supplemented with 0.1% v/v Tween-20 (PanReac AppliChem). Detection was performed by means of chemiluminescence (Bio-Rad ChemiDoc) and protein levels were quantified with Image Lab software (RRID: SCR_014210, version 6.0.1, Bio-Rad, https://www.bio-rad.com/en-be/product/image-lab-software?ID=KRE6P5E8Z). A detailed protocol can be found at: https://doi.org/10.17504/protocols.io.e6nvwdqz2lmk/v1.
14C-labeled polyamine uptake – This protocol is based on a previous publication [15], with minor modifications. Briefly, 70% confluent SH-SY5Y cells in a 12-well plate were incubated (30 min, 37 °C) either with 5 µM 14C-labeled spermine (3139-50 µCi, ARC) or with a mixture of 5 µM 14C-spermine and 100 µM unlabeled spermine in cell culture medium. The medium was subsequently aspirated and cells were washed twice with ice-cold PBS (without Ca2+ and Mg2+). Next, the cells were lysed by incubation in 200 µl radio-immunoprecipitation assay buffer (10 min, room temperature) (Thermo Fisher Scientific) before scraping. The cell lysate was added to scintillation vials filled with 7 ml EcoLite Liquid Scintillation Cocktail (01882475-CF, MP Biomedicals). Thereafter, the wells were washed with 200 µl ice-cold PBS (without Ca2+ and Mg2+), which was added to the accompanying scintillation vial. Finally, 14C radioactivity in counts-per-minute (CPM) was measured with liquid scintillation counting (TRI-CARB 4910TR V Liquid Scintillation Counter, PerkinElmer). A detailed protocol can be found at: https://doi.org/10.17504/protocols.io.j8nlkorm5v5r/v1.
Microsome collection - SH-SY5Y cells overexpressing wild-type or mutant ATP13A2 were seeded in 500 cm2 plates. Once they reached 70–80% confluency, cells were collected. Next, cells were lysed by resuspending the cell pellet in 3 ml hypotonic LIS buffer (10 mM Tris.HCl pH 7.5, 0.5 mM MgCl2.6H2O, 1 mM DTT, 1x SigmaFast protease inhibitors) (S8830, Merck), which was incubated on ice for 15 min. The suspension was transferred to a Dounce homogenizer and 60 up-and-down strokes were applied, followed by addition of 3 ml 1 M solution (0.5 M sucrose, 10 mM Tris.HCl pH 7.3, 40 µM CaCl2, 1 mM DTT, 1x SigmaFast protease inhibitors) and another 30 up-and-down strokes. The nuclear (1000 x g, 10 min, 4 °C), mitochondrial-lysosomal (12 000 x g, 20 min, 4 °C), and microsomal fractions (140,000 x g, 35 min, 4 °C) were then collected. Fractions were suspended in 0.25 M sucrose with 1x SigmaFast protease inhibitors. A detailed protocol can be found at: https://doi.org/10.17504/protocols.io.5qpvo3w7dv4o/v1.
ADP-Glo assay - ATPase activity was assessed using a commercially available luminescence assay (ADP-Glo Max assay, V7002, Promega) according to manufacturer’s instructions. A reaction mixture (final volume 25 µl) was made in a 96-well plate and contained 50 mM MOPS-KOH (pH 7), 100 mM KCl, 11 mM MgCl2, 1 mM DTT, 0.1 mg/ml DDM, 5 µg microsomes (1:2 ratio DDM: microsomes) and various concentrations of polyamines. The microsomes were collected from SH-SY5Y cells overexpressing ATP13A2 (wild-type or mutants). Next, the reaction was incubated (20 min, 37 °C followed by 10 min, room temperature) following the addition of 5 mM ATP and terminated by adding 25 µl of ADP-Glo Reagent. The 96-well plate was subsequently incubated (40 min, room temperature), followed by the addition of 50 µl of ADP-Glo Detection Reagent. After 60 min, luminescence was detected using the Bio Tek plate reader. A detailed protocol can be found at: https://doi.org/10.17504/protocols.io.3byl4q1x8vo5/v1.
RNA collection and qPCR - RNA was isolated using the NucleoSpin RNA plus kit (Macherey-Nagel) following manufacturer’s instructions. RNA concentration and purity were determined using a Nanodrop spectrometer (Thermo Fisher Scientific). 5 µg RNA was then converted to cDNA using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). A 96-well plate was prepared with a ten-fold dilution of each cDNA sample in duplicates (with a volume of 5 µl cDNA per well), to which the following reaction mixture was added (volumes are given per well): 10 µl SYBR Green master mix (Roche), 1 µl of 5 µM forward primer, 1 µl of 5 µM reverse primer, and 3 µl distilled water. The cDNA was replaced by an equivalent volume of distilled water for the negative controls. Primers targeting the ATP13A2 gene were 5’-CATGGCTCTGTACAGCCTGA-3’ (forward) and 5’ CTCATGAGCACTGCCACTGT-3’ (reverse). Primers targeting the β-actin gene were 5’-CACTGAGCGAGGCTACAGCTT-3’ (forward) and 5’-TTGATGTCGCGCACGATTT-3’ (reverse). The qPCR read-out was performed, in which the reaction was initiated at 95 °C for 10 min, followed by 50 cycles at 95 °C for 10 s and 55 °C for 30 s, and ended at 95 °C for 1 min. A melting curve was determined from 55 to 95 °C. A detailed protocol can be found at: https://doi.org/10.17504/protocols.io.14egn3ymzl5d/v1.
Literature search
PubMed and Embase searches were completed in April 2023 (and updated in December 2023) using the terms “kufor-rakeb”, “kufor rakeb”, “KRPPD”, ”parkinson disease-9”, “parkinson disease 9”, “PARK9”, “pallido-pyramidal degeneration with supranuclear upgaze paresis and dementia”, “ATP13A2”, or “1p36.13”, in combination with “schizophrenia”, “psychosis”, “psychotic”, “hallucinations”, “delusions”, “paranoia”, “antipsychotic”, or “neuroleptic”. The Pubmed search yielded nine results and the Embase search yielded 26. After duplicates were removed, 33 results remained. All results were manually reviewed, including their reference lists, for additional relevant articles. OMIM was also reviewed. Only articles that described the use of antipsychotic medications in individuals with Kufor-Rakeb syndrome were included in the review. The search process is outlined in Fig. 2.

Literature Search flow diagram for antipsychotic use in Kufor-Rakeb syndrome
Results
Biochemical analysis of the ATP13A2 variant
To functionally characterize the novel ATP13A2 variant described in this study, we generated SH-SY5Y neuroblastoma cells overexpressing either wild-type ATP13A2, a catalytically dead variant that is defective in auto-phosphorylation (D508N, functioning as a negative control) or the Pro652_Glu653del variant. Cells overexpressing Pro652_Glu653del presented a significantly lower uptake of 14C-labeled spermine as compared to cells overexpressing wild-type ATP13A2 (Fig. 3A), which offers a read-out for ATP13A2’s transport activity. This correlated well with the spermine-dependent ATPase activity in the membrane fractions of these cell lines. The ATPase activity of the Pro652_Glu653del protein was comparable to the D508N loss-of-function mutant pointing to complete loss of activity (Fig. 3B). Whereas wild-type and D508N ATP13A2 colocalized with CD63, a late endolysosomal marker, the Pro652_Glu653del variant displayed a mesh-like pattern suggesting mislocalization in the endoplasmic reticulum (Fig. 3C). We also analyzed the expression level, and found that Pro652_Glu653del mRNA levels were significantly higher than wild-type (Fig. 3D), demonstrating successful transduction. In contrast, the Pro652_Glu653del variant exhibited a lower protein expression than wild-type or D508N ATP13A2 (Fig. 3E), suggesting that protein stability may be impaired. In conclusion, in line with other previously described KRS variants [5], the Pro652_Glu653del variant exhibits full loss-of-function, which can be explained by a combination of protein instability, mislocalization, and inactivity. Our biochemical analysis indicates that in a homozygous context, this variant may be disease causing.

Functional characterization of the Pro652_Glu653del ATP13A2 variant. SH-SY5Y neuroblastoma cells either non-transduced (nts) or stably transduced with constructs encoding for wild-type ATP13A2 (WT), a catalytically dead variant (D508N), or the Pro652_Glu653del (P652_E653del) ATP13A2 variant were used to analyze (A) cellular polyamine uptake potential by scintillation counting following a 30 min incubation with 14C-labeled spermine. CPMA, counts-per-minute (n = 3). (B) Spermine-induced ATPase activity was determined in isolated microsomes (n = 3). Fitting was performed using the non-linear allosteric sigmoidal. (C) Colocalization of ATP13A2 (in green) with the late endolysosomal marker CD63 (in red) was analyzed by immunofluorescence (n = 3). Scale bar, 5 μm. Individual and merged channels are shown for the boxed areas. ATP13A2 mRNA (n = 3) (D) and protein (n = 3) (E) levels in the SH-SY5Y cells were assessed via qPCR and immunoblotting and qPCR, respectively. *, p < 0.05; **, p < 0.01; ****, p < 0.0001; ns, non-significant versus WT (A, D) or nts (E); one-sample t-test. Graphs were created with GraphPad Prism (RRID: SCR_002798, Prism - GraphPad)
Literature search
Although psychotic symptoms have often been described in KRS, the effectiveness of antipsychotic therapy has only been described for six individuals [16,17,18,19,20,21,22]; specifically with respect to olanzapine [20], aripiprazole [21, 22], quetiapine [16, 20], clozapine [19], and thioridazine (which unsurprisingly led to a worsening of motor symptoms) [18]. Although at least a partial and/or temporarily sustained therapeutic response was reported in all cases, it is not clear that any individuals achieved a full and enduring remission of psychotic symptoms. The one possible exception is the individual described by Balint et al. [21] who eventually “remained off antipsychotics without paranoid outbreaks”; however, no further details were provided, and there is also no mention of whether her visual hallucinations recurred. Additional details pertaining to the individuals’ psychiatric phenotypes and response to treatment, as well as to their particular ATP13A2 variants, are outlined in Table 1. Developmental and neurological history information is presented in Table 2. Additionally, narrative summaries for each case are provided in Supplemental Table 1 (outlining their psychiatric phenotypes) and Supplemental Table 2 (outlining their developmental and neurological phenotypes). Five additional articles alluded to the use of antipsychotic medication but did not provide any information regarding treatment response [23,24,25,26,27]. The clinical details pertaining to the psychiatric aspects of these additional five cases are outlined in Supplemental Table 3.
Discussion
Interestingly, various antipsychotic medications known to cause EPS were used in most previous case reports, despite KRS being a form of early-onset parkinsonism. In some cases, even typical or first generation antipsychotics, which are particularly problematic in this respect, were used. Although McNiel-Gauthier et al. [22] suggested that low dose aripiprazole was effective without definitive evidence of medication-related EPS in their patient, this was presumably speculative given his progressive motor dysfunction.
Quetiapine and clozapine, which are commonly used in PD when antipsychotic therapy is deemed necessary, were only used in a few cases. Specifically, quetiapine was only used in three patients [16, 19, 20], and its effectiveness and/or impact on motor functioning were only vaguely described by Di Fonzo et al. [16] and Pietrzak et al. [20]. Moreover, the concurrent use of levodopa in both cases [16, 20], as well as the use of olanzapine in one [20], further confounds interpretation of quetiapine’s effectiveness and tolerability in these patients. Clozapine (at a very low dose) has only been used in one published case to date [19]. Although this patient’s hallucinations reportedly improved, it is not clear that their symptoms fully remitted.
Here, we describe a KRS patient carrying a novel homozygous variant in ATP13A2, which was demonstrated to cause a complete loss of protein activity, in accordance with previously characterized ATP13A2 KRS-causing variants [3, 5, 9,10,11,12,13]. Importantly, this is the first report of a patient with KRS whose psychotic symptoms remitted with quetiapine monotherapy. Notably however, Di Fonzo et al. [16] and Pietrzak et al. [20] both utilized a total daily dose of 50 mg, whereas we ultimately had to target a higher dose (400 mg total daily dose). Although quetiapine has so far been well tolerated in our patient, we cannot be certain that it has not contributed to his ongoing motor symptoms, as olanzapine and quetiapine were cross-titrated. That is, at no point was he off of antipsychotic medication entirely; however, he and his family observed significant improvement with respect to his motor functioning following the switch to quetiapine. It should also be noted that at the time of publication, our patient was not yet on dopaminergic therapy of any kind, and it is possible that quetiapine may prove less effective should concurrent levodopa treatment eventually be required. Similarly, quetiapine may prove to be less effective and/or well tolerated as his illness progresses.
Although controlled trials in neurodegenerative parkinsonian disorders have been disappointing [28], quetiapine is nonetheless commonly prescribed for PD psychosis [29] and similarly represents a reasonable option in patients with KRS, given its side effect profile and ease of use. Although clozapine has more evidence in the treatment of PD psychosis [30], we did not pursue a clozapine trial given that our patient’s psychotic symptoms remitted with quetiapine therapy. Although clozapine can also be considered, particularly when psychotic symptoms are treatment resistant and provided no medical contraindications exist, its use is limited by the possibility of rare but potentially serious side effects and the related need for regular blood monitoring. It is also worth noting that no previous articles have described the use of pimavanserin in KRS-associated psychosis, despite pimavanserin being the only FDA approved medication for the treatment of PD psychosis in the United States [30]. This was not an option for our patient, as it is currently not available in our region.
Conclusion
This report characterizes a homozygous novel loss-of-function ATP13A2 variant in an individual with KRS. This is also the first description of psychotic symptoms remitting in response to quetiapine monotherapy in KRS. Given its lower propensity to cause EPS, quetiapine should be considered in the management of KRS-associated psychosis when antipsychotic therapy is deemed necessary.
Data availability
All datasets generated or analyzed in this study can be found through the Zenodo repository (doi:10.5281/zenodo.10600748). All experimental protocols are shared via protocols.io. For the purpose of open access, the author has applied a CC BY 4.0 public copyright license to all Author Accepted Manuscripts arising from this submission.
References
Gregory A, Hayflick S (1993) Neurodegeneration with Brain Iron Accumulation Disorders Overview. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A (eds) GeneReviews((R)). Seattle (WA)
Yang X, Xu Y (2014) Mutations in the ATP13A2 gene and parkinsonism: a preliminary review. Biomed Res Int 2014(371256). https://doi.org/10.1155/2014/371256
Estrada-Cuzcano A, Martin S, Chamova T, Synofzik M, Timmann D, Holemans T, Andreeva A, Reichbauer J, De Rycke R, Chang DI, van Veen S, Samuel J, Schols L, Poppel T, Mollerup Sorensen D, Asselbergh B, Klein C, Zuchner S, Jordanova A, Vangheluwe P, Tournev I, Schule R (2017) Loss-of-function mutations in the ATP13A2/PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). Brain 140:287-305.10.1093/brain/aww307
Azfar M, van Veen S, Houdou M, Hamouda NN, Eggermont J, Vangheluwe P (2022) P5B-ATPases in the mammalian polyamine transport system and their role in disease. Biochim Biophys Acta Mol Cell Res 1869:119354.10.1016/j.bbamcr.2022.119354
van Veen S, Martin S, Van den Haute C, Benoy V, Lyons J, Vanhoutte R, Kahler JP, Decuypere JP, Gelders G, Lambie E, Zielich J, Swinnen JV, Annaert W, Agostinis P, Ghesquiere B, Verhelst S, Baekelandt V, Eggermont J, Vangheluwe P (2020) ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 578:419-424.10.1038/s41586-020-1968-7
Vrijsen S, Houdou M, Cascalho A, Eggermont J, Vangheluwe P (2023) Polyamines in Parkinson’s Disease: balancing between neurotoxicity and neuroprotection. Annu Rev Biochem 92:435–464. https://doi.org/10.1146/annurev-biochem-071322-021330
Fiori LM, Turecki G (2008) Implication of the polyamine system in mental disorders. J Psychiatry Neurosci 33:102–110
Vrijsen S, Besora-Casals L, van Veen S, Zielich J, Van den Haute C, Hamouda NN, Fischer C, Ghesquiere B, Tournev I, Agostinis P, Baekelandt V, Eggermont J, Lambie E, Martin S, Vangheluwe P (2020) ATP13A2-mediated endo-lysosomal polyamine export counters mitochondrial oxidative stress. Proc Natl Acad Sci U S A 117:31198–31207. https://doi.org/10.1073/pnas.1922342117
Ning YP, Kanai K, Tomiyama H, Li Y, Funayama M, Yoshino H, Sato S, Asahina M, Kuwabara S, Takeda A, Hattori T, Mizuno Y, Hattori N (2008) PARK9-linked parkinsonism in eastern Asia: mutation detection in ATP13A2 and clinical phenotype. Neurology 70:1491-1493.10.1212/01.wnl.0000310427.72236.68
Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E (2012) Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A 109:9611–9616. https://doi.org/10.1073/pnas.1112368109
Grunewald A, Arns B, Seibler P, Rakovic A, Munchau A, Ramirez A, Sue CM, Klein C (2012) ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol Aging 33:1843 e1841-1847.10.1016/j.neurobiolaging.2011.12.035
Podhajska A, Musso A, Trancikova A, Stafa K, Moser R, Sonnay S, Glauser L, Moore DJ (2012) Common pathogenic effects of missense mutations in the P-type ATPase ATP13A2 (PARK9) associated with early-onset parkinsonism. PLoS One 7:e39942.10.1371/journal.pone.0039942
Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D (2012) Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J Neurosci 32:4240–4246. https://doi.org/10.1523/JNEUROSCI.5575-11.2012
Martin S, van Veen S, Holemans T, Demirsoy S, van den Haute C, Baekelandt V, Agostinis P, Eggermont J, Vangheluwe P (2016) Protection against Mitochondrial and Metal Toxicity Depends on Functional Lipid Binding Sites in ATP13A2. Parkinsons Dis 2016:9531917.10.1155/2016/9531917
Houdou M, Jacobs N, Coene J, Azfar M, Vanhoutte R, Van den Haute C, Eggermont J, Daniels V, Verhelst SHL, Vangheluwe P (2023) Novel Green Fluorescent Polyamines to Analyze ATP13A2 and ATP13A3 Activity in the Mammalian Polyamine Transport System. Biomolecules 1310.3390/biom13020337
Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto G, Fabbrini G, Marconi R, Fincati E, Abbruzzese G, Marini P, Squitieri F, Horstink MW, Montagna P, Libera AD, Stocchi F, Goldwurm S, Ferreira JJ, Meco G, Martignoni E, Lopiano L, Jardim LB, Oostra BA, Barbosa ER, Italian Parkinson Genetics N, Bonifati V (2007) ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 68:1557-1562.10.1212/01.wnl.0000260963.08711.08
Chien HF, Rodriguez RD, Bonifati V, Nitrini R, Pasqualucci CA, Gelpi E, Barbosa ER (2021) Neuropathologic Findings in a Patient With Juvenile-Onset Levodopa-Responsive Parkinsonism Due to ATP13A2 Mutation. Neurology 97:763-766.10.1212/WNL.0000000000012705
Behrens MI, Bruggemann N, Chana P, Venegas P, Kagi M, Parrao T, Orellana P, Garrido C, Rojas CV, Hauke J, Hahnen E, Gonzalez R, Seleme N, Fernandez V, Schmidt A, Binkofski F, Kompf D, Kubisch C, Hagenah J, Klein C, Ramirez A (2010) Clinical spectrum of Kufor-Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Mov Disord 25:1929-1937.10.1002/mds.22996
Abbas MM, Govindappa ST, Sheerin UM, Bhatia KP, Muthane UB (2017) Exome Sequencing Identifies a Novel Homozygous Missense ATP13A2 Mutation. Mov Disord Clin Pract 4:132-135.10.1002/mdc3.12353
Pietrzak A, Badura-Stronka M, Kangas-Kontio T, Felczak P, Kozubski W, Latos-Bielenska A, Wierzba-Bobrowicz T, Florczak-Wyspianska J (2019) Clinical and ultrastructural findings in an ataxic variant of Kufor-Rakeb syndrome. Folia Neuropathol 57:285-294.10.5114/fn.2019.88459
Balint B, Damasio J, Magrinelli F, Guerreiro R, Bras J, Bhatia KP (2020) Psychiatric Manifestations of ATP13A2 Mutations. Mov Disord Clin Pract 7:838-841.10.1002/mdc3.13034
McNeil-Gauthier AL, Brais B, Rouleau G, Anoja N, Ducharme S (2019) Successful treatment of psychosis in a patient with Kufor-Rakeb syndrome with low dose aripiprazole: a case report. Neurocase 25:133-137.10.1080/13554794.2019.1625928
Williams DR, Hadeed A, al-Din AS, Wreikat AL, Lees AJ (2005) Kufor Rakeb disease: autosomal recessive, levodopa-responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia. Mov Disord 20:1264-1271.10.1002/mds.20511
Schneider SA, Paisan-Ruiz C, Quinn NP, Lees AJ, Houlden H, Hardy J, Bhatia KP (2010) ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov Disord 25:979-984.10.1002/mds.22947
Estiar MA, Leveille E, Spiegelman D, Dupre N, Trempe JF, Rouleau GA, Gan-Or Z (2020) Clinical and genetic analysis of ATP13A2 in hereditary spastic paraplegia expands the phenotype. Mol Genet Genomic Med 8:e1052.10.1002/mgg3.1052
Odake Y, Koh K, Takiyama Y, Ishiura H, Tsuji S, Yamada M, Yoshita M (2020) Identification of a novel mutation in ATP13A2 associated with a complicated form of hereditary spastic paraplegia. Neurol Genet 6:e514.10.1212/NXG.0000000000000514
Satolli S, Di Fonzo A, Zanobio M, Pezzullo G, De Micco R (2023) Kufor Rakeb syndrome without gaze palsy and pyramidal signs due to novel ATP13A2 mutations. Neurol Sci 44:3723-3725.10.1007/s10072-023-06899-2
Desmarais P, Massoud F, Filion J, Nguyen QD, Bajsarowicz P (2016) Quetiapine for Psychosis in Parkinson Disease and Neurodegenerative Parkinsonian Disorders: A Systematic Review. J Geriatr Psychiatry Neurol 29:227-236.10.1177/0891988716640378
Friedman JH (2018) Pharmacological interventions for psychosis in Parkinson’s disease patients. Expert Opin Pharmacother 19:499-505.10.1080/14656566.2018.1445721
Rissardo JP, Durante I, Sharon I, Fornari Caprara AL (2022) Pimavanserin and Parkinson’s Disease Psychosis: A Narrative Review. Brain Sci 1210.3390/brainsci12101286
Acknowledgements
Not applicable.
Funding
The authors acknowledge the financial support of the Fonds Wetenschappelijk Onderzoek (FWO) Flanders (G094219N to P.V. and 1S88419N to S.V.). P.V., S.V., R.A.E.A., C.V.D.H. and M.H. are funded by the joint efforts of The Michael J. Fox Foundation for Parkinson’s Research (MJFF) and the Aligning Science Across Parkinson’s (ASAP) initiative. MJFF administers the grant ASAP-000458 on behalf of ASAP and itself.
Author information
Authors and Affiliations
Contributions
MC (psychiatrist), GM (psychiatrist), JS (neurologist), and PYA (clinical geneticist) were all involved in the assessment and treatment of the patient and contributed to the development and writing of the manuscript. PV, SV, RAEA, MH, and CVDH were all involved in the design and completion of the biochemical analysis of the genetic variant and also collectively contributed to the writing of the corresponding portions of the manuscript.
Corresponding author
Ethics declarations
Ethical approval
The patient’s legal guardian has provided written informed consent for the publication of this report, and the patient himself has assented. KU Leuven Ethical Commission provided approval to work with human SHSY5Y cell lines.
Declaration of Generative AI and AI-assisted technologies in the writing process
No AI or AI-assisted technologies were used in the preparation or writing of this manuscript.
Submission declaration
A longer version of this manuscript has previously been posted on the preprint server medRxiv (https://www.medrxiv.org/content/10.1101/2024.01.12.23300401v2). It has otherwise not been published elsewhere and is not under consideration for publication elsewhere.
Competing interests
MC has no conflicts of interest directly relevant to this paper. MC is a co-investigator for a RCT in generalized anxiety disorder sponsored by Sunovion and Sumitomo, and a study physician for a RCT in major depressive disorder partially funded by Otsuka (part of CAN-BIND). He also provides psychiatric consultation for the ATLAS study (Biogen). He has not received any money from these companies for this work. PV is involved in a drug discovery program for ATP13A2 agonists for Parkinson’s disease.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Mark Ainsley Colijn and Stephanie Vrijsen shared first authorship.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Colijn, M.A., Vrijsen, S., Au, P.Y.B. et al. Kufor-Rakeb syndrome-associated psychosis: a novel loss-of-function ATP13A2 variant and response to antipsychotic therapy. Neurogenetics (2024). https://doi.org/10.1007/s10048-024-00767-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10048-024-00767-7