
Abstract
Mutations in FUS and TBK1 often cause aggressive early-onset amyotrophic lateral sclerosis (ALS) or a late-onset ALS and/or frontotemporal dementia (FTD) phenotype, respectively. Co-occurrence of mutations in two or more Mendelian ALS/FTD genes has been repeatedly reported. However, little is known how two pathogenic ALS/FTD mutations in the same patient interact to shape the final phenotype. We screened 28 ALS patients with a known FUS mutation by whole-exome sequencing and targeted evaluation for mutations in other known ALS genes followed by genotype–phenotype correlation analysis of FUS/TBK1 double-mutant patients. We report on new and summarize previously published FUS and TBK1 double-mutant ALS/FTD patients and their families. We found that, within a family, mutations in FUS cause ALS while TBK1 single mutations are observed in FTD patients. FUS/TBK1 double mutations manifested as ALS and without a manifest difference regarding age at onset and disease duration when compared to FUS single-mutant individuals. In conclusion, TBK1 and FUS variants do not seem to interact in a simple additive way. Rather, the phenotype of FUS/TBK1 double-mutant patients appears to be dominated by the FUS mutation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating motor neuron disease. Its heritability is estimated to be in the range of 30–60%. Overall, mutations in more than 30 genes have been associated with ALS and the clinically and genetically overlapping disease frontotemporal dementia (FTD) in the last decades [1, 2]. Five percent of the patients present with a positive family history for ALS/FTD compatible with an autosomal-dominant and very rarely autosomal-recessive or X-linked dominant Mendelian mode of inheritance. Furthermore, an oligogenic causation in familial and sporadic ALS/FTD is increasingly appreciated [3, 4]. Co-occurrence of mutations in two or more ALS disease genes in the same patient may increase the penetrance of some ALS-associated mutations that have a low effect size. In genetic mouse models, the presence of two different ALS mutations can modify the disease phenotype in a complex manner: We and others have previously shown that expression of mutant SOD1 or TARDBP/TDP-43 in mice interacts with heterozygous loss-of-function (LoF) mutations in TBK1 to alter the motor neuron disease phenotype in mice [5,6,7]. However, little is known about the possible interaction of two different ALS mutations in humans. One reason for this is the rarity of identical double mutations in patients, and consequently the small number of patients whose phenotype could be compared to patients with respective single mutations. Moreover, very rarely a pedigree has been published of affected family members carrying mutations in either one or both genes, what would allow phenotypic comparisons on a similar genetic background.
Mutations in the ALS causing genes FUS and TBK1 are observed in about 4% and 2% of familial ALS (FALS) cases in Germany, respectively [8]. Mutations in the RNA-binding protein FUS can cause ALS and FTD in very rare instances. ALS-causing mutations in FUS lead to a nucleocytoplasmic redistribution and cytoplasmic aggregation of FUS protein [9, 10]. The heterozygous ALS- and FTD-causing mutations in TBK1 usually lead to a loss of function of one TBK1 allele and have been suggested to impair the cellular role of TBK1 in autophagy and glial immune responses [5]. Thus, both genes act most likely at an upstream position in different cellular pathways. It is plausible to hypothesize that the presence of mutations in both genes in the same patient may result in an enhanced penetrance or a synergistic exaggeration of the clinical manifestation. Indeed, we and others have previously described several ALS patients with simultaneous mutations in FUS and TBK1 [11, 12]. In this paper, we compare the co-segregation of genotypes and phenotypes in two families in which mutations in TBK1 and FUS occur separately or in combination and compare them with previously reported and newly identified TBK1/FUS double-mutant patients.
Results
Screening for patients with variants in FUS and TBK1
Based on previous findings of co-occurrence of FUS and TBK1 mutations in familial ALS patients [11, 12], we screened 28 ALS patients (21 FALS index patients and seven affected relatives) with a known FUS mutation by whole-exome sequencing and targeted evaluation for mutations in other known ALS genes. A prior screening for C9ORF72 had been unsuspicious in these patients. Indeed, we found two additional familial ALS patients with FUS/TBK1 double mutations: FUS c.1540A > G; p.R514G together with TBK1 c.1328_1331del; p.I443Nfs*3 and FUS c.1562G > A; p.R521H in combination with TBK1 c.1522C > A; p.L508I (patients A and C in Table 1). In addition, we found FUS mutations to co-occur with missense variants in ANXA11 (c.772C > T; p.V258M) and SETX (c.2113A > C; p.I705L) in two patients (see Supplementary Information and Supplementary Table 1).
Overview of previously published FUS/TBK1 double-mutant families
Lattante and colleagues have previously performed genetic testing of 413 Italian ALS patients (32 FALS and 381 SALS) using panel analysis of 32 known ALS/FTD genes [12]. This screen revealed three SALS patients carrying both a TBK1 variant and a variant in FUS. All three FUS variants were located in the 3’ UTR in FUS (patients E–G in Table 1). Furthermore, de Majo et al. described a FUS c.1561C > T; p.R521C together with a TBK1 c.1073G > A; p.R358H mutation in two first-degree relatives (patient D in Table 1) [13]. In a cohort of 252 whole-exome sequenced German FALS patients, we had previously identified another pedigree with a TBK1 LoF variant (c.555 T > A; p.Y185*) co-occurring with a FUS missense mutation (c.1570A > G; p.R524G) (Fig. 1B; family B in Table 1) [11]. Thus, to our knowledge, at least five index patients have been previously reported who carry both a TBK1 and a FUS variant. Together with the new data from this paper, we summarize different TBK1 and FUS variants found to co-occur in a total of seven index patients, of which four carry a diagnosis of familial ALS. However, the evidence for pathogenicity of the respective mutations differs between these patients. Only in two of the seven FUS/TBK1 double-mutant index patients (patients A and B in Table 1) pathogenicity can be regarded to be proven for both the FUS and the TBK1 variants (patients A and B), as outlined in detail in Table 1 and the Supplementary Information.

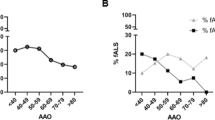
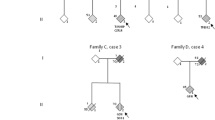
Genotype–phenotype analysis of FUS/TBK1 double-mutant patients. A, B Pedigrees of the two FALS families with co-occurrence of mutations in TBK1 and FUS. Arrows indicate index patients. Age at onset (in years) and duration of disease (in months) are indicated next to or below each patient. C, D Bar graphs showing age of onset and disease duration (time since disease onset until death or tracheostomy) of FUS/TBK1 double-mutant ALS patients (consisting of patients A II.4, B II.3, B II.5, and B III.2 in Table 2) compared to a large international cohort of FUS-ALS patients
FUS mutations define the ALS phenotype of FUS/TBK1 double patients
Our further analysis focuses on the pedigrees with patients with evidence for causality in both FUS and TBK1, specifically the families of index patients A and B. We performed Sanger sequencing of both genes in available members of the two families and identified two additional FUS/TBK1 double-mutant ALS patients. This resulted in a group of four double-mutant FUS/TBK1 ALS patients consisting of patients A II.4, B II.3, B II.5, and B III.2 (see Table 2). Compared to an international cohort of 188 FUS-ALS patients [14], this group had a similar mean age at onset of ALS of 42 ± 3.5 (SD) vs. 39.97 ± 16.2 years (p = 0.8) and a mean disease duration (time since disease onset until death or tracheostomy) of 57.8 ± 53.3 vs. 41.9 ± 43.6 months (p = 0.48) (Fig. 1C and D). However, we point out that the disease characteristics of the cohort in Naumann et al. are prominently influenced by FUS mutations that are more aggressive than the mutations p.R514G and p.R524G so that this comparison is admittedly suboptimal. The disease onset in the FUS/TBK1 double-mutant patients was spinal or bulbar (Table 2). Double-mutant patients did not exhibit comorbid FTD. Although the number of FUS/TBK1 double-mutant patients is low, the present results suggest that a combination of pathogenic variants in FUS and TBK1 does not exacerbate the ALS phenotype compared to pathogenic FUS variants only.
To gain insight into the phenotypic manifestations of FUS/TBK1 double variation on a similar genetic background, we compared single- and double-mutant members of families A and B (Table 1). As shown by Fig. 1, the single mutations in FUS and TBK1 co-segregate with ALS or FTD, respectively. In the symptomatic members of both families, the TBK1 variants alone precipitate a FTD phenotype. By contrast, FUS variants alone, or in combination with the TBK1 variants, cause ALS without FTD, while an obvious additive or synergistic effect of FUS and TBK1 variants on the phenotype is not observed in these two families.
In family A, the likely FUS mutant father (I.1) shows an unusually late ALS onset, while his FUS/TBK1 double-mutant son (II.4) had a very early onset of ALS. In family B, both the obligate FUS mutant father (I.2) and the FUS/TBK1 double-mutant children (II.3 and II.5) and grandson (III.2) show similarly early onsets of ALS (Fig. 1B). Unfortunately, DNA from the family B member II.4 was unavailable. While the disease duration of the ALS patient I.2 with the obligate isolated FUS mutation is not known, his children with FUS/TBK1 double mutations (II.3 and II.5) both displayed a relatively long disease course if compared to the mean survival time of FUS-ALS patients (Fig. 1D). The FUS/TBK1 double-mutant index patient II.5 of family B has even survived for more than 13 years after ALS onset. By contrast, the grandson (III.2) shows an aggressive disease course.
Discussion
In this analysis, we extend the previous knowledge about FUS/TBK1 double-mutant patients by whole-exome sequencing of additional 28 patients with FUS mutation. Overall, we report and summarize eight patients with rare variants in both FUS and TBK1, weigh the evidence for their pathogenicity, and study the phenotype of double-mutant compared to single-mutant ALS patients. Importantly, we report several single- and double-mutant ALS patients in two families, i.e., on a comparable genetic background.
The rationale to perform the extended screening for TBK1 variants in FUS mutation carriers was based on the previous description of patients with concomitant variants in FUS and TBK1 [11, 12]. Our study revealed additional double-mutant individuals, and both TBK1 and FUS belong to the less frequently mutated ALS disease genes. Nevertheless, larger patient numbers will be required to show that mutations in both genes occur more frequently than expected by chance. If not a by chance finding, the penetrance of mutations in FUS and TBK1 could be increased when mutations in both genes co-occur in one individual, thus increasing the probability to be detected in a patient. The 72-year-old asymptomatic FUS mutation carrier in family B (II.2) may be regarded as support for a lower penetrance in single-mutant patients.
In our two FUS/ALS families, the combination of TBK1 and FUS variants led to early-onset ALS without FTD comorbidity, while the TBK1 variants alone caused FTD and the isolated FUS variants precipitated early-onset ALS. Possibly, FUS/TBK1 double mutation carriers with early ALS do not survive to the later age at which TBK1-associated FTD usually starts. Consequently, the FUS mutations principally shape the disease phenotype in FUS/TBK1 patients.
A surprising finding of our study is that our initial “double-hit” hypothesis — that the presence of two mutations in the same patient would result in an exacerbated disease through summated neurotoxicity — did not prove true. Comparing the disease characteristics of the group of double FUS/TBK1 mutant ALS patients of this study (four patients) with a large cohort of mostly single FUS mutant ALS patients, we found that the age of onset and disease duration were unaltered by the combination of FUS and TBK1 mutations. Considering the apparently complementary pathomechanisms of FUS and TBK1 mutations (in particular impaired protein quality control due to TBK1 haploinsufficiency and FUS proteinopathy as a consequence of FUS mutations), this is a remarkable finding. Nevertheless, a larger cohort of FUS/TBK1 double-mutant patients together with mechanistic studies using FUS/TBK1 double-mutant disease models are warranted to corroborate the findings of our study.
In conclusion, the neurotoxic effects of TBK1 and FUS mutations do not seem to add up in a simple way in patients. Rather, the phenotype of FUS/TBK1 double-mutant patients appears to be dominated by the FUS mutation. These insights may have relevance also for the design of gene-specific therapies for both single- and double-mutant patients.
Methods
Patients and ethics statements
All ALS patients were diagnosed according to the EFNS Consensus criteria [15]. The study was approved by the medical ethical review boards of the universities of Ulm and Umea. With informed written consent and in accordance with the Declaration of Helsinki, EDTA blood samples were drawn from controls, ALS patients, and their unaffected relatives during visits at the Neurology departments of the University Hospitals of Ulm and Umea. DNA was extracted from EDTA blood samples according to standard procedures.
Genotyping of patients for SOD1 and C9ORF72 mutations
Mutations in SOD1 and C9ORF72 were excluded prior to exome sequencing of familial ALS cases as described before [11].
Whole-exome sequencing
Whole-exome sequencing, read mapping, and variant calling were performed on HiSeq2000/2500 systems (Illumina) as described previously [11].
Statistics
For comparison of two groups, the unpaired two-tailed Student’s t test was used. Data are presented as means ± SEM in bar graphs. Statistical significance was reported by the p value of the statistical test procedures and was assessed as significant (*, P < 0.05), strongly significant (**, P < 0.01), or highly significant (***, P < 0.001; ****, P < 0.0001). All statistical analyses were performed with Prism software (version 9.1.1; GraphPad Software).
Data availability
Available upon request.
Code availability
Not applicable.
Change history
16 February 2022
Open Access funding enabled and organized by Projekt DEAL has been added as funding.
References
White MA, Sreedharan J (2016) Amyotrophic lateral sclerosis. Curr Opin Neurol 29:557–564. https://doi.org/10.1097/WCO.0000000000000367
Brenner D, Weishaupt JH (2019) Update on amyotrophic lateral sclerosis genetics. Curr Opin Neurol 32:735–739. https://doi.org/10.1097/WCO.0000000000000737
Van Blitterswijk M, Van Es MA, Hennekam EAM et al (2012) Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet 21:3776–3784. https://doi.org/10.1093/hmg/dds199
Nguyen HP, Van Broeckhoven C, van der Zee J (2018) ALS genes in the genomic era and their implications for FTD. Trends Genet 34:404–423
Brenner D, Sieverding K, Bruno C et al (2019) Heterozygous Tbk1 loss has opposing effects in early and late stages of ALS in mice. J Exp Med jem.20180729. https://doi.org/10.1084/jem.20180729
Gerbino V, Kaunga E, Ye J et al (2020) The loss of TBK1 kinase activity in motor neurons or in all cell types differentially impacts ALS disease progression in SOD1 mice. Neuron 106:789-805.e5. https://doi.org/10.1016/j.neuron.2020.03.005
Sieverding K, Ulmer J, Bruno C et al (2021) Hemizygous deletion of Tbk1 worsens neuromuscular junction pathology in TDP-43G298S transgenic mice. Exp Neurol 335. https://doi.org/10.1016/j.expneurol.2020.113496
Müller K, Brenner D, Weydt P et al (2018) Comprehensive analysis of the mutation spectrum in 301 German ALS families. J Neurol Neurosurg Psychiatry jnnp-2017-317611. https://doi.org/10.1136/jnnp-2017-317611
Kwiatkowski TJ, Bosco DA, LeClerc AL et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science (80- ) 323:1205–1208. https://doi.org/10.1126/science.1166066
Vance C, Rogelj B, Hortobágyi T et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science (80- ) 323:1208–1211. https://doi.org/10.1126/science.1165942
Freischmidt A, Wieland T, Richter B et al (2015) Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci 18:631–636. https://doi.org/10.1038/nn.4000
Lattante S, Doronzio PN, Marangi G et al (2019) Coexistence of variants in TBK1 and in other ALS-related genes elucidates an oligogenic model of pathogenesis in sporadic ALS. Neurobiol Aging 84:239.e9-239.e14. https://doi.org/10.1016/j.neurobiolaging.2019.03.010
de Majo M, Topp SD, Smith BN et al (2018) ALS-associated missense and nonsense TBK1 mutations can both cause loss of kinase function. Neurobiol Aging 71:266.e1-266.e10. https://doi.org/10.1016/j.neurobiolaging.2018.06.015
Naumann M, Peikert K, Günther R et al (2019) Phenotypes and malignancy risk of different FUS mutations in genetic amyotrophic lateral sclerosis. Ann Clin Transl Neurol 6:2384–2394. https://doi.org/10.1002/acn3.50930
Andersen PM, Abrahams S, Borasio GD et al (2012) EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)—revised report of an EFNS task force. Eur J Neurol 19:360–375. https://doi.org/10.1111/j.1468-1331.2011.03501.x
Pozzi L, Valenza F, Mosca L et al (2017) TBK1 mutations in Italian patients with amyotrophic lateral sclerosis: genetic and functional characterization. J Neurol Neurosurg Psychiatry 88:869–875. https://doi.org/10.1136/jnnp-2017-316174
Acknowledgements
We are indebted to the patients and their families for their participation in this project.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported in whole or in part by grants of the Medical Faculty Mannheim of the University of Heidelberg (Mannheim Advanced Clinician Scientist [MACS] program), the Medical Faculty of the University of Ulm (Clinician Scientist Baustein program), the Charcot Foundation for ALS research (Ulm), the German society for patients with muscular diseases (DGM), and the German Federal Ministry of Education and Research (JPND ‘STRENGTH’ consortium [01ED1408]; JPND ‘PreFrontAls’ [01ED1512]; the German Network for ALS Research MND-NET [01GM1103A]).
Author information
Authors and Affiliations
Contributions
Conception and design of the study: D.B., J.H.W. Acquisition and analysis of data and revision of the manuscript: D.B., K.M., S.L., R.Y., A.K., A.F., A.C.L., P.M.A., J.H.W.
Corresponding author
Ethics declarations
Ethics approval/consent to participate/consent for publication
The study was approved by the medical ethical review boards of the universities of Ulm and Umea. Written informed consent for participation and publication was obtained in accordance with institutional requirements and the study fulfilled the principles of the Declaration of Helsinki.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brenner, D., Müller, K., Lattante, S. et al. FUS mutations dominate TBK1 mutations in FUS/TBK1 double-mutant ALS/FTD pedigrees. Neurogenetics 23, 59–65 (2022). https://doi.org/10.1007/s10048-021-00671-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-021-00671-4