
Abstract
Spastic ataxias are rare neurogenetic disorders involving spinocerebellar and pyramidal tracts. Many genes are involved. Among them, CAPN1, when mutated, is responsible for a complex inherited form of spastic paraplegia (SPG76). We report the largest published series of 21 novel patients with nine new CAPN1 disease-causing variants and their clinical characteristics from two European university hospitals (Paris and Stockholm). After a formal clinical examination, causative variants were identified by next-generation sequencing and confirmed by Sanger sequencing. CAPN1 variants are a rare cause (~ 1.4%) of young-adult-onset spastic ataxia; however, together with all published cases, they allowed us to better describe the clinical and genetic spectra of this form. Truncating variants are the most frequent, and missense variants lead to earlier age at onset in favor of an additional deleterious effect. Cerebellar ataxia with cerebellar atrophy, dysarthria and lower limb weakness are often associated with spasticity. We also suggest that cognitive impairment and depression should be assessed specifically in the follow-up of SPG76 cases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Spastic ataxias are rare neurodegenerative genetic disorders involving spinocerebellar and pyramidal tracts. They belong to the clinical spectrum of hereditary spastic paraplegias (HSPs) and hereditary cerebellar ataxias (HCAs). Variants in more than a hundred genes have been implicated in their etiology [1, 2]. All classical transmission modes have been described.
CAPN1 variants were initially associated with an autosomal recessive (AR) form of complex HSP (SPG76) or spastic ataxia in 2016 [3, 4]. Since then, an increasing number of patients have been described (n > 50). CAPN1 encodes the calpain-1 protein, a calcium-activated intracellular proteinase, with numerous biological roles, including in synaptic plasticity and neuroprotection [5].
Gene panel screening and exome or genome sequencing in two European university hospitals (Paris and Stockholm) allowed us to diagnose 21 novel cases carrying 13 CAPN1 causative variants, the largest series reported so far. We report here the clinical and genetic characteristics of this cohort, including nine novel variants. Furthermore, we explored genotype-phenotype correlations based on our findings and a review of all previously published cases.
Materials and methods
Patients were clinically examined according to described clinical criteria (https://spatax.wordpress.com/downloads/). Disease severity was measured using the SPATAX disability scale (0: no functional handicap; 1: no functional handicap but signs at examination; 2: mild, able to run, walking unlimited; 3: moderate, unable to run, limited walking without aid; 4: severe, walking with one stick; 5: walking with two sticks; 6: unable to walk, requiring wheelchair; 7: confined to bed). The study was approved by the Stockholm and Paris-Necker Ethics Committees. All patients signed informed consent for genetic analyses, which were conducted in accordance with the relevant national ethical rules. Their DNA collected from blood cells was used for genome sequencing (six probands), exome sequencing (two probands), or targeted gene panel sequencing (five probands with four among 182 probands previously excluded for pathogenic variants in 65 HSP genes [6]). Standard procedures were used for next-generation sequencing, sequence alignment and filtering, as described elsewhere [6, 7]. All variants were confirmed by Sanger sequencing, and their co-segregation with the disease was also verified in other family members when DNA was available. Novel variants have been submitted to the ClinVar database.
The literature review was done from all CAPN1-mutated cases reported in PubMed or ClinVar before October 2020. The search was done in PubMed with ‘CAPN1’ as the search term, and the papers reporting a description of cases harboring variants in CAPN1 were kept for the review.
Mean and standard deviation were used to describe continuous quantitative variables, and median and interquartile range (iqr) were used for discrete quantitative variables. Student’s t test was used to compare means of onset age, disease duration and handicap that followed a normal distribution, as assessed visually and by Shapiro-Wilk test. Chi2 test was used to compare distributions of clinical features. A p value < 0.05 was considered as statistically significant.
Results
Case series
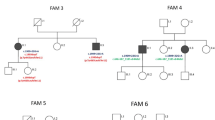
We diagnosed a series of 21 cases of spastic ataxia with CAPN1 causative variants from 13 new families (Fig. 1). Clinical and genetic data are summarized in Table 1 and full clinical data are shown in Supplementary Table 1.

Pedigrees of CAPN1 families. Affected filled in black. Squares for men, circles for women, lozenges for men or women, small circles if stillborn. Numbers in squares and circles denote the number of people with similar characteristics to simplify the figure. Below the symbols are indicated the anonymous DNA sample identifier. Arrows show the probands
Thirteen different disease-causing variants were found, and nine of these are reported here for the first time: c.254G>A (p.Trp85*), c.623G>A (p.Gly208Asp), c.1697dup (p.Leu566Phefs*7), and c.1969G>T (p.Glu657*) at the homozygous state, and c.1005G>A (p.Trp335*), c.1129_1133del (p.Arg377Glufs*25), c.1165+1G>A (p.?), c.1341G>C (splice site variant), and c.1418_1419del (p.Arg473Leufs*53) at the compound heterozygous state. These novel variants were not reported in gnomAD [8]. In total, half of the cases were from consanguineous unions, explaining a high homozygosity rate of 71% (15 cases). All our variants were truncating except one (8%). The only non-truncating damaging missense variant, c.623G>A (p.Gly208Asp), affected a highly conserved residue and was predicted to alter the protein function by all commonly used prediction scores (CADD, MutationTaster2, PolyPhen-2, and SIFT) [9,10,11,12]. The c.1341G>C variant was considered as truncating as it was highly predicted to affect the splicing by deletion of the splicing donor site at the end of exon 11 (prediction with MaxEntScan − 100.0%, NNSPLICE − 94.6% and SSF − 16.2%) [13,14,15]. The study of mRNA from white blood cells of the proband identified multiple abnormal transcripts compared with controls, one of which could be sequenced and shown to result from partial retention of the following intron, leading to a frame shift and a premature stop codon: p.Pro449Argfs*17.
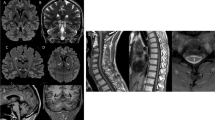
The patients originated from various geographical locations: 11 from Western Europe, six from North Africa, one from Eastern Europe and three from Iraq. The sex ratio was 1.33 (p > 0.05). Mean age at onset of the disease was 25.1 ± 8.8 years, and mean age at examination was 41.0 ± 11.4 years. Disease severity using the SPATAX disability scale showed variable levels of handicap, with a median score of 3 (iqr 3–4) after a mean disease duration of 16.2 ± 9.7 years. Most of the patients had walking difficulties (17/21) with two at score 1, two at 2, seven at 3, five at 4 and five at 5. Lower limb spasticity, extensor plantar reflex and lower limb hyperreflexia were present in all patients whereas upper limb spasticity was found in 10 of the 18 cases with available data (56%). Lower limb weakness was present in 16/20 cases (80%). In 60% of cases (12/20), dysarthria was noted and was described as spastic (n = 5), bulbar (n = 3) or cerebellar (n = 4). Cerebellar ataxia was observed in 10/19 cases (53%). Other frequent features were sphincter dysfunction (primarily bladder) in 10/18 (56%), sensory deficit (always in deep modality and also superficial in one case) in 7/19 (37%), saccadic pursuit in 3/18 (17%) and dysphagia in 4/19 (21%). A single case among 19 patients with available examination (5%; patient FSP-1225-8) had extrapyramidal signs with the development of orofacial dystonia and hypokinesia during the follow-up, without any exposure to neuroleptics. Three affected relatives from family FSP-1225 presented an early bilateral cataract starting in their forties, which was also present in individual 11 who had no spastic ataxia. No eye fundus anomalies were detected. Skeletal abnormalities were reported in 6/19 cases (32%) as scoliosis, pes cavus, hollow foot, C2–3 fusion with supernumerary hemivertebra and unspecified dental abnormalities. Interestingly, a cognitive impairment was observed in two sisters (family SR37-12333, 2/19 or 11%) after three decades of disease evolution. In addition, 7/17 patients (41%) had psychiatric manifestations with mood disorders that manifested as depression, post-traumatic stress disorder or bipolar disorder. Epilepsy was not reported. Brain MRI was abnormal in 4/15 cases (27%) with cerebellar atrophy, clinically observed as ataxia in three of them. Spine MRI showed upper spine atrophy in 1/9 cases (11%). Neuropathy was confirmed by electroneuromyography (ENMG) in a single case (9%; patient FSP-327-771).
The clinical variability was evident between family members and between some affected cases sharing the same causative variant in different families. For example, the age at onset ranged from 18 to 36 years in family FSP-1225 (patients homozygous for c.1697dup) and it ranged from 15 to 30 years for the recurrent c.1176G>A variant in the two families carrying it at the homozygous state. Genotype to phenotype correlations could be found with neither the position nor the consequence of the variant in our series of 21 patients.
A prospective follow-up is given in Table 1 with two clinical examinations for patient FSP-1225-8 at 35 and 41 years old. It showed that the evolution of the disease course was progressive with a pure spastic paraparesis at onset complicated over time by cerebellar ataxia, orofacial dystonia and increased disability.
Discussion
We report 13 CAPN1 variants in 21 patients from 13 families, nine being novel causative variants. While some families were found through exome, genome or targeted panel sequencing in large cohorts of unselected patients with various phenotypes, four were identified in a series of 182 HSP patients screened for pathogenic variants in known HSP genes [6], allowing us to estimate the relative frequency of CAPN1 variants in HSP patients. These 182 cases were from a series of 292 cases, 37.7% of whom were found to have mutations in known HSP genes (unpublished data), suggesting that CAPN1 variants account for 1.4% of our European continental HSP patients. This is higher than in a CAPN1 screening of 107 AR or sporadic HSP cases and 54 HCA Chinese patients, where a frequency of 0.6% was reported [16], but is in line with the frequency of 1/47 (2.1%) paediatric-onset HSP patients [17]. Due to the recent establishment of CAPN1 as a disease gene, more cases could well be found in undiagnosed patients, particularly as the diagnosis process is evolving from targeted analysis of a few known disease genes to high-throughput analyses of the entire genome.
Literature review
There are 34 previously published pathogenic CAPN1 variants in 37 families or 62 cases in PubMed or ClinVar (Supplementary Table 2) [3, 4, 16, 18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33]. One of these variants was reported in ClinVar without clinical data (c.1153C>T) [VCV000489122.2], and we confirmed this variant as disease-causing in one of our cases. Consanguinity is frequent, accounting for 63% of all known families. Our new cases slightly increased the sex ratio to 0.76 from the previous ratio of 0.61, but the difference remains non-significant. Nevertheless, females are more frequently affected by complex forms (p < 0.01) as already reported [19]. All known variants are distributed throughout the whole gene (Fig. 2). Most variants were private, affecting a single family except for the most frequent, c.1176G>A, present in seven families, c.1605+5G>A found in four families, c.188dup and c.759+1G>A both identified in three families, and c.618_619del, c.853C>T, c.1142C>T, c.1153C>T, c.1493C>T, c.1534C>T and c.1969G>T, each detected in two families. A common ancestor could be hypothesized for the most frequent variant, c.1176G>A, since some mutated cases shared common origins, such as Brazil or Portugal (the others were from Turkey and France). In contrast, there was no known common origin for the four families carrying the c.1605+5G>A variant, which affects a CG doublet, known as a potential mutational hotspot and thus making a founder effect less likely. The various types of variants occurring in this gene are nonsense, missense, frameshift, and splice site variants. Truncating variants are frequent and represent 29 (67%) of the 43 CAPN1 causative variants now known with our supplementary series or also 40 (80%) of the 50 known families. The number of truncating variants could be slightly overestimated due to predicted splicing variants that may not have truncating consequences, as is the case for c.337+1G>A (p.Leu112ins9) [4]. Indeed, the reported splicing variants have rarely been functionally studied for their consequences (n = 7/11 without mRNA analysis).

Calpain-1 with its 3 functional domains and all reported pathogenic variants (new in bold; truncating variants localized with a lozenge whereas a simple line for non-truncating variants. Below the protein are the corresponding CAPN1 coding exons)
As reported recently, the frequency of truncating variants did not differ between pure and complex forms [19] but, interestingly, we found that they are associated with a later onset than non-truncating missense variants (mean of 26.9 ± 7.2 vs 17.5 ± 8.1 years, p < 0.001, Fig. 3). We therefore hypothesize that the pathogenic missense variants in CAPN1 could cause additional negative effects, although their smaller number (n = 16 cases) makes conclusions difficult especially as the handicap does not differ after the same disease duration (p = 0.3) of 12.1 ± 9.0 years for the missense variants and 14.8 ± 9.0 years for the truncating variants. The case with the earliest onset, at 1 year, supports this hypothesis of an increased severity of missense variants; but this patient was reported to carry three missense variants (c.221G>A (p.Gly74Asp) + c.911C>T (p.Thr304Met) + c.1418G>T (p.Arg473Leu)), all predicted to affect protein structure [32]. The second earliest cases, two patients with onset at 13 years, were also caused by missense variants [26, 30]. In truncating variants, the earliest onset was 14 years in c.1153C>T (p.Arg385*) in one of our families (11264-SA).

Clinical summary of the 83 CAPN1-mutated cases reported in the literature, including our 21 cases (number at the end of each line indicates the number of cases with available data)
Pure HSPs are estimated at 36% (Clopper-Pearson confidence interval 0.95, 26–47%) of all known CAPN1 cases, but missing reported clinical data could lead to this being overestimated and the progression of the neurodegeneration could complicate the initial pure clinical presentation in some patients. The distribution of associated clinical features of CAPN1 variants from all known cases is summarized in Fig. 3 to show the global clinical signature of CAPN1-mutated cases. Lower limb spasticity is a constant feature, and spasticity often extends to the upper limbs (68% or 28/41 cases with available data). Thereafter, cerebellar ataxia, dysarthria and lower limb weakness are the most frequent signs. Dysarthria is more frequent in AR than in autosomal dominant (AD) forms of HSP. It is found in 62% of SPG11 cases, one of the most frequent AR-HSPs, and up to 83% in DDHD2, a rare, severe HSP [34]. Dysarthria is also frequent in CAPN1 patients, involving 42/61 (69%) of cases. CAPN1 dysarthria results from the combination of pyramidal, bulbar and cerebellar involvement. Skeletal anomalies are reported in up to 16/37 (43%) of cases, and are therefore more frequent than deep sensory deficit (12/53, 23%). Brain and spine MRI may show atrophy, especially of cerebellum, brainstem and upper cervical cord (17/49 patients, or 35%, had abnormal MRI). Finally, the intra-familial and intra-variant phenotypic variability suggests individual factors modulating the variant consequences.
To reflect its clinical variability, the name ‘CAPN1-associated neurodegeneration’ has been proposed to describe the disease in patients carrying disease-causing variants in this gene [20]. It is now clear that some HSP genes are shared with many other neurodegenerative disorders [1]. However, screening in populations displaying early-onset Parkinson’s disease and youth-onset amyotrophic lateral sclerosis did not find any CAPN1 pathogenic variants [16]. Notably, some of the CAPN1 consanguineous cases also carry other pathogenic variants in DYSF or MEFV, so the phenotype in these cases may result from a synergistic effect of these two genes [28, 33]. The patient with CAPN1 and DYSF pathogenic variants did indeed have a severe muscle weakness attributed to the muscular dystrophy.
In contrast to previous reports, we point out the importance of psychological assessment, with 7/17 cases (41%) described here being prone to depression. Depression is rarely reported and probably rarely assessed in spastic ataxia except for SCA1 and SCA3 [35]. We think that mood disorders are underestimated in HSPs; indeed, a previous study found that a half of HSP patients were subject to depression [36]. Whether this is a consequence or part of the pathological process is unclear as most published series lack this clinical information. In addition, cognitive impairment seems to occur during the evolution of the disease after two decades, as we have seen in two of our oldest cases. Such features have previously been reported only once, after 23 years of evolution and in the form of a frontotemporal dementia-like presentation associated with the same variant as one of our cases (c. 1176G>A) [25].
CAPN1 causative variants result especially in deficiency of calpain-1-mediated cleavage of PH domain and leucine rich repeat protein phosphatase 1 (PHLPP1) [4]. This deficiency is suggested to cause inhibition of the Akt pro-survival pathway and, then, neurodegeneration. Activation of the Akt-pathway or inhibition of PHLPP1 is a therapeutic approach effective in mice to prevent neurodegeneration caused by CAPN1 disease-causing variants [4]. This may open avenues of therapeutic intervention in the future.
Conclusion
We described a large series of 21 patients from 13 families with CAPN1 pathogenic variants causing various degrees of spastic ataxia, including nine novel variants.
CAPN1 disease-causing variants are a rare cause of spastic ataxia of young adult onset, often involving consanguineous families. Truncating variants are the most frequent gene alterations. Interestingly, we found with the analysis of all the previously reported cases and our cases that the non-truncating missense variants are associated with an earlier age at onset than the truncating ones, which argues in favour of a stronger deleterious effect of the mutant protein compared with the truncating forms, which are probably degraded through non-sense-mediated mRNA decay. The associated clinical presentation is variable. Dysarthria and spasticity are the core features with mild cerebellar signs. Other signs are rarer, but some cases with cognitive impairment and/or depression indicate that these signs should be assessed specifically in the follow-up of spastic ataxia with CAPN1 causative variants.
Many cases remain to be diagnosed and more data should be collected to know more about the natural history of this form.
Data availability
All CAPN1 variants identified in this study have been declared to the ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar).
References
Parodi L, Coarelli G, Stevanin G, Brice A, Durr A (2018) Hereditary ataxias and paraparesias: clinical and genetic update. Cur Op Neurol 31:462–471
Synofzik M, Schüle R (2017) Overcoming the divide between ataxias and spastic paraplegias: shared phenotypes, genes, and pathways. Mov Disord 32:332–345
Gan-Or Z, Bouslam N, Birouk N, Lissouba A, Chambers DB, Vérièpe J, Androschuk A, Laurent SB, Rochefort D, Spiegelman D, Dionne-Laporte A, Szuto A, Liao M, Figlewicz DA, Bouhouche A, Benomar A, Yahyaoui M, Ouazzani R, Yoon G, Dupré N, Suchowersky O, Bolduc FV, Parker JA, Dion PA, Drapeau P, Rouleau GA, Ouled Amar Bencheikh B (2016) Mutations in CAPN1 cause autosomal-recessive hereditary spastic paraplegia. Am J Hum Genet 98:1038–1046
Wang Y, Hersheson J, Lopez D, Hammer M, Liu Y, Lee KH, Pinto V, Seinfeld J, Wiethoff S, Sun J, Amouri R, Hentati F, Baudry N, Tran J, Singleton AB, Coutelier M, Brice A, Stevanin G, Durr A, Bi X, Houlden H, Baudry M (2016) Defects in the CAPN1 gene result in alterations in cerebellar development and cerebellar ataxia in mice and humans. Cell Rep 16:79–91
Baudry M, Bi X (2016) Calpain-1 and Calpain-2: the Yin and Yang of synaptic plasticity and neurodegeneration. Trends in Neurosciences 39:235–245
Morais S, Raymond L, Mairey M, Coutinho P, Brandão E, Ribeiro P, Loureiro JL, Sequeiros J, Brice A, Alonso I, Stevanin G (2017) Massive sequencing of 70 genes reveals a myriad of missing genes or mechanisms to be uncovered in hereditary spastic paraplegias. Eur J Hum Genet 25:1217–1228
Coutelier M, Hammer MB, Stevanin G, Monin ML, Davoine CS, Mochel F, Labauge P, Ewenczyk C, Ding J, Gibbs JR, Hannequin D, Melki J, Toutain A, Laugel V, Forlani S, Charles P, Broussolle E, Thobois S, Afenjar A, Anheim M, Calvas P, Castelnovo G, de Broucker T, Vidailhet M, Moulignier A, Ghnassia RT, Tallaksen C, Mignot C, Goizet C, le Ber I, Ollagnon-Roman E, Pouget J, Brice A, Singleton A, Durr A, for the Spastic Paraplegia and Ataxia Network (2018) Efficacy of exome-targeted capture sequencing to detect mutations in known cerebellar ataxia genes. JAMA Neurol 75:591–599
Genome Aggregation Database Consortium, Karczewski KJ, Francioli LC et al (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434–443
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47:D886–D894
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11:361–362
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249
Sim N-L, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC (2012) SIFT web server: predicting effects of amino acid substitutions on proteins. Nucl Acids Res 40:W452–W457
Yeo G, Burge CB (2004) Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11:377–394
Reese MG, Eeckman FH, Kulp D et al (1997) Improved splice site detection in genie. J Comput Biol 4:311–323
Shapiro MB, Senapathy P (1987) RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucl Acids Res 15:7155–7174
Xia Z-C, Liu Z-H, Zhou X-X, Liu Z, Wang JL, Hu ZM, Tan JQ, Shen L, Jiang H, Tang BS, Lei LF (2020) Mutation analysis of CAPN1 in Chinese populations with spastic paraplegia and related neurodegenerative diseases. J Neurol Sci 411:116691
Travaglini L, Aiello C, Stregapede F, D’Amico A, Alesi V, Ciolfi A, Bruselles A, Catteruccia M, Pizzi S, Zanni G, Loddo S, Barresi S, Vasco G, Tartaglia M, Bertini E, Nicita F (2018) The impact of next-generation sequencing on the diagnosis of pediatric-onset hereditary spastic paraplegias: new genotype-phenotype correlations for rare HSP-related genes. Neurogenet 19:111–121
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, Karapetyan K, Katz K, Liu C, Maddipatla Z, Malheiro A, McDaniel K, Ovetsky M, Riley G, Zhou G, Holmes JB, Kattman BL, Maglott DR (2018) ClinVar: improving access to variant interpretations and supporting evidence. Nucl Acids Res 46:D1062–D1067
Lai L-L, Chen Y-J, Li Y-L, Lin XH, Wang MW, Dong EL, Wang N, Chen WJ, Lin X (2020) Novel CAPN1 mutations extend the phenotypic heterogeneity in combined spastic paraplegia and ataxia. Ann Clin Transl Neurol 7:1862–1869
Rahimi Bidgoli MM, Javanparast L, Rohani M, et al (2020) CAPN1 and hereditary spastic paraplegia: a novel variant in an Iranian family and overview of the genotype-phenotype correlation. Int J Neurosci :1–17.
Hengel H, Buchert R, Sturm M, Haack TB, Schelling Y, Mahajnah M, Sharkia R, Azem A, Balousha G, Ghanem Z, Falana M, Balousha O, Ayesh S, Keimer R, Deigendesch W, Zaidan J, Marzouqa H, Bauer P, Schöls L (2020) First-line exome sequencing in Palestinian and Israeli Arabs with neurological disorders is efficient and facilitates disease gene discovery. Eur J Hum Genet 28:1034–1043
Garcia-Berlanga JE, Moscovich M, Palacios IJ, et al (2019) CAPN1 variants as cause of hereditary spastic paraplegia type 76. Case Rep Neurol Med :7615605.
Wei Q, Dong H-L, Pan L-Y, Chen CX, Yan YT, Wang RM, Li HF, Liu ZJ, Tao QQ, Wu ZY (2019) Clinical features and genetic spectrum in Chinese patients with recessive hereditary spastic paraplegia. Transl Neurodegener 8:19
Cotti Piccinelli S, Bassi MT, Citterio A et al (2019) A Novel CAPN1 mutation causes a pure hereditary spastic paraplegia in an Italian family. Front Neurol 10:580
Souza PVS, Silva LHL, Badia BML, Farias IB, Pinto WBVR, Oliveira ASB (2019) SPG76: An extremely rare hereditary spastic paraplegia with a new expanding complicated phenotype. Rev Neurol (Paris) 175:572–574
Kim A, Kumar KR, Davis RL, Mallawaarachchi AC, Gayevskiy V, Minoche AE, Walls Z, Kim HJ, Jang M, Cowley MJ, Choi JH, Shin C, Sue CM, Jeon B (2019) Increased diagnostic yield of spastic paraplegia with or without cerebellar ataxia through whole-genome sequencing. Cerebellum 18:781–790
Peng F, Sun Y-M, Quan C, Wang J, Wu JJ (2019) Two novel homozygous mutations of CAPN1 in Chinese patients with hereditary spastic paraplegia and literatures review. Orphanet J Rare Dis 14:83
Shetty A, Gan-Or Z, Ashtiani S, Ruskey JA, van de Warrenburg B, Wassenberg T, Kamsteeg EJ, Rouleau GA, Suchowersky O (2019) CAPN1 mutations: expanding the CAPN1-related phenotype: From hereditary spastic paraparesis to spastic ataxia. Eur J Med Genet 62:103605
Melo US, Freua F, Lynch DS, Ripa BD, Tenorio RB, Saute JAM, de Souza Leite F, Kitajima J, Houlden H, Zatz M, Kok F (2018) Clinical aspects of hereditary spastic paraplegia 76 and novel CAPN1 mutations. Clin Genet 94:482–483
Lambe J, Monaghan B, Munteanu T, Redmond J (2018) CAPN1 mutations broadening the hereditary spastic paraplegia/spinocerebellar ataxia phenotype. Pract Neurol 18:369–372
Kocoglu C, Gundogdu A, Kocaman G, Kahraman-Koytak P, Uluc K, Kiziltan G, Caglayan AO, Bilguvar K, Vural A, Basak AN (2018) Homozygous CAPN1 mutations causing a spastic-ataxia phenotype in 2 families. Neurol Genet 4:e218
Travaglini L, Bellacchio E, Aiello C, Pro S, Bertini E, Nicita F (2017) Expanding the clinical phenotype of CAPN1-associated mutations: a new case with congenital-onset pure spastic paraplegia. J Neurol Sci 378:210–212
Tadic V, Klein C, Hinrichs F, Münchau A, Lohmann K, Brüggemann N (2017) CAPN1 mutations are associated with a syndrome of combined spasticity and ataxia. J Neurol 264:1008–1010
Erfanian Omidvar M, Torkamandi S, Rezaei S, Alipoor B, Omrani MD, Darvish H, Ghaedi H (2019) Genotype–phenotype associations in hereditary spastic paraplegia: a systematic review and meta-analysis on 13,570 patients. J Neurol. Epub ahead of print (19 November 2019). https://doi.org/10.1007/s00415-019-09633-1
Schmitz-Hübsch T, Coudert M, Tezenas du Montcel S, Giunti P, Labrum R, Dürr A, Ribai P, Charles P, Linnemann C, Schöls L, Rakowicz M, Rola R, Zdzienicka E, Fancellu R, Mariotti C, Baliko L, Melegh B, Filla A, Salvatore E, van de Warrenburg BPC, Szymanski S, Infante J, Timmann D, Boesch S, Depondt C, Kang JS, Schulz JB, Klopstock T, Lossnitzer N, Löwe B, Frick C, Rottländer D, Schlaepfer TE, Klockgether T (2011) Depression comorbidity in spinocerebellar ataxia. Mov Disord 26:870–876
Vahter L, Braschinsky M, Haldre S, Gross-Paju K (2009) The prevalence of depression in hereditary spastic paraplegia. Clin Rehabil 23:857–861
Acknowledgments
The authors are grateful to the patients and their relatives participating in this study, and to Dr Rabea Zemmouri who provided clinical data. The ICM I-Genseq facility and DNA and Cell Bank were also crucial for most of the studies performed by the authors.
Funding
This study was funded in part by the Fundação para a Ciência e Tecnologia (FCT, grant FCT-ANR/BEX-GMG/0008/2013, to IA) and by the French National Agency for Research (ANR, SPATAX-QUEST, to GS), the European Union HORIZON2020 program (SOLVE-RD, to GS), the ERARE program (NEUROLIPID and PREPARE_ataxia, to GS) and the VERUM Foundation (to VH and GS) and benefited from the program “Investissements d’Avenir” (ANR-11-INBS-0011 – NeurATRIS: Translational Research Infrastructure for Biotherapies in Neurosciences, to the ICM institute). SM was the recipient of a fellowship (no. SFRH/BD/87189/2012) from the FCT, and MC was the recipient of an Aspirant fellowship from the Fonds de la Recherche Scientifique (FNRS). JLM received an “Année Recherche” grant from Rouen University Hospital.
Author information
Authors and Affiliations
Contributions
CF, GC, MK, RR, EP, MT, FT, KL, JPA, DN, AB, RP, JLL, CG, AD and MaP performed the recruitment and clinical evaluations of patients. JLM, RV, VH, GB, SM, LP, MC and MeP performed the genetic analyses. PS, IA, ELG, GV, MaP and GS supervised the work. JLM and GS wrote the first draft of the manuscript. All authors read the manuscript, contributed to the manuscript revision and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Consent to participate (including appropriate statements)
All patients gave written informed consent for genetic testing and study related procedures.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Table 1
Full clinical table of our new CAPN1-mutated cases (PDF 625 kb)
Supplementary Table 2
Literature review of all published CAPN1-mutated cases (until 10/2020) (PDF 785 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Méreaux, JL., Firanescu, C., Coarelli, G. et al. Increasing involvement of CAPN1 variants in spastic ataxias and phenotype-genotype correlations. Neurogenetics 22, 71–79 (2021). https://doi.org/10.1007/s10048-020-00633-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-020-00633-2