
Abstract
Li2S–P2S5–LiI-type solid electrolytes, such as Li4PS4I, Li7P2S8I, and Li10P3S12I, are promising candidates for anode layers in all-solid-state batteries because of their high ionic conductivity and stability toward Li anodes. However, few studies have been conducted on their detailed local structure and particle surface state. In this study, Li7P2S8I (Li2S: P2S5:LiI = 3:1:1) solid electrolytes as the chemical composition were synthesized by mechanical milling and liquid-phase shaking, and their local structures were analyzed by transmission electron microscopy. The particle surface states were analyzed by X-ray photoelectron spectroscopy, high-energy X-ray scattering measurements, and neutron total scattering experiments. The results showed that Li7P2S8I solid electrolytes are composed of nanocrystals, such as Li4PS4I, LiI, Li10P3S12I and an amorphous area as the main region, indicating that the crystalline components alone do not form ionic conductive pathways, with both the amorphous and crystalline regions contributing to the high ionic conductivity. Moreover, the ionic conductivity of the crystalline/amorphous interface of the glass-ceramic was higher than that of the Li2S–P2S5–LiI glass. Finally, an organic-solvent-derived stable surface layer, which was detected in the liquid-phase shaking sample, served as one of the factors that contributed to its high stability (which surpassed that of the mechanically milled sample) toward lithium anodes. We expect these findings to enable the effective harnessing of particle surface states to develop enhanced sulfide solid electrolytes.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
All-solid-state Li batteries have attracted significant attention owing to their several advantages over conventional organic liquid electrolyte batteries, such as higher safety, higher energy density, superior rate performance, and wider operational temperature range. Unlike traditional Li-ion batteries, which use organic liquid electrolytes, all-solid-state batteries use solid electrolytes to facilitate the movement of Li ions between the cathode and anode layers. The choice of solid electrolyte is therefore crucial for the overall performance and safety of batteries.
Solid electrolytes used in all-solid-state batteries can be classified into two types based on their composition. The most widely studied inorganic solid electrolytes are oxide- and sulfide-based types [1]. In particular, sulfide-based solid electrolytes such as Li3PS4 [2], Li6PS5Cl [3], Li7P3S11 [4], Li10GeP2S12 [5], and Li9.54Si1.74P1.44S11.7Cl0.3 [6] are reported to exhibit superior ionic conductivities at room temperature compared to oxide-types; Li9.54[Si0.6Ge0.4]1.74P1.44S11.1Br0.3O0.6, in particular, is a new sulfide solid electrolyte that has exhibited the highest ionic conductivity reported to date of 32 mS at room temperature [7]. These electrolytes also have soft crystal lattices, owing to which they deform easily, which presses the electrolyte particles together and connects them, thus facilitating ion transport across the grain boundaries [8, 9]. Solid electrolytes are used not only in the separator layer but also in the cathode and anode composite layers; therefore, it is necessary for them to be stable toward the cathode or anode while exhibiting a high ionic conductivity.
Several previous studies indicate that halogen-doped sulfide solid-state electrolytes play a significant role in enhancing the cycling performance of Li metal solid-state batteries [10,11,12,13]. In particular, Li2S–P2S5–LiI-type solid electrolytes, such as Li4PS4I, Li7P2S8I, and Li10P3S12I, are also promising candidates for use in the anode layers in all-solid-state batteries owing to their high ionic conductivities of 2.3–9.0 mS at room temperature and high stability toward Li anodes [14,15,16,17,18]. Li2S–P2S5–LiI-type solid electrolytes are mainly synthesized by mechanical milling. However, the liquid-phase synthesis of Li7P2S8I using acetonitrile [19], ethyl propionate [20], and so on is being considered a viable method for its large-scale production.
Li2S–P2S5–LiI-type solid electrolytes with a chemical composition of Li7P2S8I (Li2S: P2S5:LiI = 3:1:1) have been extensively studied and are known to form glass-ceramics composed of crystalline and amorphous regions [21, 22]. A previous report on Li2S–P2S5 glass-ceramics suggested that the interfacial layer contains a highly Li-ion-conducting pathway that enhances the total conductivity [23]. High-resolution transmission electron microscopy (TEM) images of Li2S–P2S5 glass-ceramics revealed the presence of nanocrystals in an amorphous region and that the crystal–amorphous region interface is the key to achieving a high ionic conductivity [24]. However, detailed local structural analyses, such as evaluation of the crystal/amorphous interface of Li7P2S8I solid electrolytes, have been rarely conducted; therefore, further investigations are required to determine the local structures. In addition, studies on the surface state of the Li7P2S8I solid-electrolyte particles and its effect on the electrochemical properties are scarce.
In this study, Li7P2S8I solid electrolytes were synthesized by mechanical milling and liquid-phase shaking, and their local structures were analyzed by TEM. The particle surface states of both synthesis routes were analyzed using X-ray photoelectron spectroscopy (XPS), high-energy X-ray scattering measurements, and neutron total scattering experiments. As a result, the local structures of the Li7P2S8I solid electrolytes, which affected ionic conduction, became apparent. Unlike the mechanically milled sample, the liquid-phase-synthesized sample showed an organic-solvent-derived stable surface layer, which contributed to the high stability of the latter toward Li anodes.
Experimental
Solid electrolyte synthesis
Li2S (99.9%; Mitsuwa Chemicals), P2S5 (99%; Merck), and LiI (99.9%; Aldrich) were mixed at a molar ratio of 3:1:1 using 4-mm zirconia balls (approximately 32 g) and solvent ethyl propionate (10 mL; Aldrich) as the solvent in a sealed 45-mL polypropylene centrifugation tube (Labcon North America). Ethyl propionate was employed because it has an appropriate boiling point (approximately 99 °C) and has also been listed by the US Department of Health and Human Services Food and Drug Administration (FDA) as a non-toxic solvent (class 3) [25]. A suspension was obtained by shaking the mixture in the tube at 1500 rpm for 3 h under a dry Ar atmosphere [26]. After the suspension was initially evacuated at room temperature for 1 h, the temperature was increased to 70 °C for 0.5 h and 90, 110, or 130 °C for 2 h under vacuum using a rotary vacuum pump (GHD-031 A, ULVAC). The precursor powder was pelletized (pellet diameter = 10 mm, approximately 100 mg) by uniaxial pressing under 254 MPa at 25 °C. The pellets were then placed in an SiO2 tube, to which Ar gas was supplied through the inlet and passed out through the outlet; the precursor pellets were then heated in a tube furnace developed for this study (Fig. 1). The pellets were then carefully ground using an agate mortar to obtain a Li7P2S8I solid electrolyte powder. For comparison, we also synthesized Li7P2S8I by mechanical milling in a planetary ball mill (Pulverisette 7; Fritsch) at a rotation speed of 500 rpm for 15 h followed by heat treatment at 180 °C for 3 h.

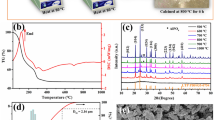
XRD patterns and temperature dependence of the ionic conductivity of the Li7P2S8I solid electrolytes synthesized by the liquid-phase shaking (LS) method [26]
Characterization
The crystal structures of the solid electrolytes were characterized by X-ray diffraction (XRD; Ultima IV and Smartlab SE, Rigaku Co., Ltd.). The samples were sealed in specialized holders (Rigaku Co., Ltd.) in an Ar-filled glove box to avoid exposure to humid air. The morphologies of the solid electrolytes were evaluated using a field-emission scanning electron microscope (FE-SEM; S-4800, Hitachi High-Tech Co., Ltd.). Synchrotron XRD patterns were obtained on the BL02B2 beamline located at the SPring-8 facility using a high-flux synchrotron X-ray source with a wavelength of 0.6199 Å [27]. Each sample was sealed in a quartz capillary tube (diameter: 0.3 mm) under vacuum, and images were acquired using a Debye–Scherrer diffraction camera. Field-emission transmission electron microscopy (FE–TEM) was conducted at an accelerating voltage of 200 kV at room temperature using the JEM-2100 F (JEOL Ltd.) [28]. The samples for TEM were mounted on an amorphous carbon film supported by a Cu grid inside a glove box filled with inert Ar gas. Air exposure was avoided by using a vacuum-transfer TEM holder (Mel-Build). The microstructures of the prepared Li7P2S8I samples were evaluated based on bright-field (BF) and hollow-cone dark-field (HCDF) images and the corresponding electron diffraction (ED) patterns. The incident electron beam was tilted at a fixed angle and automatically rotated along the optical axis of the objective lens [29, 30], owing to which the diffraction spots in the ED patterns were accurately reflected in the corresponding HCDF images. The precipitated crystalline phase was identified using the “ProcessDiffraction” software package. The obtained ED patterns were converted into one-dimensional intensity profiles for matching with a powder XRD pattern database [31, 32].
Electrochemical properties
The relationship between temperature and the total conductivity (which includes the bulk and the grain boundary resistances) of the solid electrolytes was investigated from 1 MHz to 10 Hz under dry Ar flow using alternating-current electrochemical impedance spectroscopy (EIS; SI 1260, Solartron Analytical). The samples for EIS were prepared by uniaxially pressing ~ 100 mg of the samples into ~ 10-mm-diameter pellets under a pressure of 254 MPa at room temperature. The pellets were placed in a polyether ether ketone (PEEK) holder with two pieces of carbon paper as the blocking electrodes. The cells were placed under an Ar flow in a glass tube for temperature-dependent measurements. The temperature was gradually increased from room temperature to 110 °C at varying increments and held at each temperature for 1 h prior to the impedance measurement. DC polarization measurements were conducted using a charge–discharge measurement tool (BST-2004 H, Nagano) with a Li/solid electrolyte/Li symmetric cell that was prepared by attaching Li foil to both sides of the pelletized solid electrolyte; a current density of ± 0.3 mA cm–2 was applied with a charge and discharge time of 1 h.
Structure analysis
XPS measurements were used to analyze the surface states of the Li7P2S8I solid electrolytes. XPS measurements of the S 2p, P 2p, C 1s, and O 1s orbitals were performed at BL7U of the Aichi Synchrotron Radiation Center. The incident X-ray energies were 260 eV for S 2p and P 2p, and 650 eV for C 1s and O 1s. The kinetic energy was converted to binding energy before analysis using Casa XPS. The obtained spectra were calibrated using Au 4f (88 and 84 eV). High-energy X-ray total scattering measurements using synchrotron radiation were performed on the BL04B2 beamline at SPring-8 [33, 34]. The X-ray system employed an incident energy of 61.339 keV and consisted of four CdTe detectors and three Ge detectors. To enable total X-ray scattering, all samples were hermetically sealed in 2.0-mmΦ borosilicate glass capillaries (WJM-Glas/Muller GmbH) inside a glove box under an Ar atmosphere with precise dew point control. The required scattering intensities I(Q) were meticulously corrected for the background, Compton scattering, polarization, and absorption. Subsequently, the structure factor S(Q) was extracted, and the reduced pair distribution function (PDF) G(r) was obtained using the Fourier transform of S(Q) [28]. The G(r) value of ethyl propionate was simulated and stretched by multiplying the horizontal axis Q of S(Q) by the appropriate coefficient (0.905). The resulting value was used as the Fourier transform of the G(r) value of ethyl propionate to determine the difference between the Li7P2S8I solid electrolytes synthesized by mechanical milling and liquid-phase shaking. Neutron total scattering experiments were performed on Li7P2S8I solid electrolytes sealed in a V-Ni null scattering sample container (outer diameter: 3.0 mm, thickness: 0.1 mm) at 27 °C for an exposure time of 3.5 h using a NOVA neutron total scattering instrument (BL21 beamline) with a decoupled liquid hydrogen moderator using an incident flight path of 15 m and a scattered flight path of 1.2–1.3 m at the 90° detector bank (0.008 < d (= 2π/Q = λ/(2 sin θ)) < 0.63 nm) connected to a 800-kW spallation neutron source at the Japan Proton Accelerator Research Complex. Samples for all analytical experiments were prepared within an Ar-filled glove box and carefully transported to a vacuum chamber without exposure to air or moisture.
Results and discussion
Basic characterization
Previously reported XRD patterns and ionic conductivity results of Li7P2S8I solid electrolytes synthesized by liquid-phase shaking are shown in Fig. 1 [26]. The XRD peaks corresponding to the Li4PS4I phase (space group P4/nmm, conductivity = 0.1 mS cm–1 at room temperature) [35] and Li10P3S12I phase with an LGPS-type structure (conductivity = 9.0 mS cm–1 at room temperature) exhibit peak at approximately 2θ = 20°–21° [16, 36, 37]; LiI peaks appear at approximately 2θ = 26°. These results indicate that the Li7P2S8I solid electrolytes are glass-ceramics composed of Li4PS4I, Li10P3S12I with a LGPS-type structure and LiI. We then conducted synchrotron XRD measurements (Fig. S1); no clear peaks corresponding to the starting materials in the Li7P2S8I precursor powder were observed. After crystallization, peaks corresponding to Li4PS4I, the LGPS-type phase, and LiI were observed in the profiles of the Li7P2S8I solid electrolytes synthesized using the liquid-phase shaking method. This result indicated that the samples synthesized by liquid-phase shaking did not exhibit additional peaks, which were otherwise present in the XRD profiles measured in the laboratory. Conversely, the mechanically milled samples exhibited broad patterns similar to the peaks corresponding to Li10P3S12I (Fig. S2). In addition, the Li7P2S8I solid electrolyte exhibited a relatively high ionic conductivity (1.0 mS cm–1) at room temperature (Fig. 1b) that was comparable to the ionic conductivity of that synthesized by mechanical milling (3.0 mS cm–1). The difference between the ionic conductivities of the samples synthesized by liquid-phase shaking and mechanical milling can be attributed to the following mechanisms: [1] The difference in the crystal phases determined by XRD analyses. The liquid-phase shaking sample is composed of the Li4PS4I phase and LiI, whose ionic conductivity is lower than that of the Li10P3S12I phase with an LGPS-type structure. However, the mechanically milled sample contains only the Li10P3S12I phase [2]. The particle size of the liquid-phase shaking sample is approximately 1 μm [26], whereas that of the mechanically milled sample is approximately 5 μm [14]. A small particle size indicates a high concentration of grain boundaries, which may increase the grain-boundary resistance in the liquid-phase shaking sample; thus, the grain-boundary resistance in the liquid-phase sample is higher than that in the mechanically milled sample.
Local structure analysis
TEM observations of the Li7P2S8I solid electrolytes synthesized by liquid-phase shaking were conducted in different fields of view to analyze their local crystal structures (Fig. 2). While halo rings were the predominant structures observed in Region 1, both diffraction spots and halo rings were observed in Region 2 (Fig. 2a). This indicates that there were non-uniform crystalline regions in the amorphous matrix. Figure 2b shows the ED intensity profiles and corresponding HCDF images obtained from different crystalline regions of the electrolyte. The HCDF images show the spatial distribution of the Li4PS4I and LiI nanocrystals, indicated by a dotted circle and arrows, respectively; the results indicated that the Li7P2S8I glass-ceramic synthesized by liquid-phase shaking was an amorphous area, which appeared as the main region and contained small amounts of Li4PS4I and LiI nanocrystals. TEM analysis of the Li7P2S8I solid electrolytes synthesized by mechanical milling (Fig. S3) revealed similar ED patterns with halo patterns in Region 1 and diffraction spots and halo rings in other regions, as well as similar HCDF images that showed LiI nanocrystals and amorphous areas as the main regions. These results indicate that the electrolytes synthesized by both methods had similar microstructures, comprising a main amorphous region with nanocrystals; the dispersion of the nanocrystals tended to be lower in liquid-phase shaken samples than in the mechanical milled samples, though the effect of this difference on the ionic conductivity is unclear. These TEM images clearly show, for the first time, that the crystalline components alone do not form ionic conductive pathways; both the amorphous regions and crystalline components contribute to the high ionic conductivity. The crystal region and/or crystalline/amorphous interface also resulted in the Li7P2S8I glass-ceramic exhibiting a higher ionic conductivity than Li2S–P2S5–LiI glass, the ionic conductivity of which is in the order of 10–4 S/cm [38]. Another glass-ceramic, Na3PS4 glass-ceramic, exhibited high room-temperature ionic conductivity in the cubic crystal after heat treatment at 270 °C (> 10–4 S cm–1) [39] because Na vacancies were introduced instead of Na sites during the crystal growth process, which is speculated to have promoted Na ionic conduction in the Na3PS4 glass-ceramic [40]. The higher ionic conductivity of the Li7P2S8I glass-ceramic compared to that of the Li7P2S8I glass may be ascribed to the vacancies formed after the crystal growth.

TEM observation results of Li7P2S8I solid electrolyte synthesized by liquid-phase shaking. a BF image and ED patterns. b ED intensity profiles and corresponding HCDF images for crystalline regions
Lithium stability
DC polarization test of the Li7P2S8I solid electrolytes was conducted to analyze their stability toward Li. Li7P2S8I synthesized by mechanical milling exhibited a constant overvoltage of 20 mV and short circuit occurred after 20 h of DC polarization at room temperature (Fig. 3). In contrast, Li7P2S8I synthesized by liquid-phase shaking maintained a constant voltage in the range of 35–47 mV even after 1200 h of DC polarization at room temperature. Therefore, Li7P2S8I synthesized by liquid-phase shaking is more stable toward Li metal than that synthesized by mechanical milling, despite exhibiting a higher overvoltage, because of its lower ionic conductivity. Thus, Li7P2S8I fabricated via liquid-phase shaking is more suitable for all-solid-state Li-metal batteries. In the next section, we attempt to clarify why the liquid-phase shaking method imparted higher stability toward Li. The electrochemical window test results of the liquid-phase shaking sample have been reported elsewhere [20]. Except the anodic and cathodic currents generated upon the lithium deposition and dissolution at 0 V, no significant current was observed from 10 to − 0.3 V. This result demonstrated that the liquid-phase shaking sample was characterized by a wide electrochemical window.

DC polarization test results of the Li7P2S8I solid electrolytes synthesized by mechanical milling and liquid-phase shaking
Particle surface states
XPS analysis
Figure 4 shows the O 1 s, C 1 s, and S 2p XPS profiles of the Li7P2S8I solid electrolytes. The mechanically milled sample showed only a single O 1 s peak corresponding to Li2CO3 at approximately 533 eV [41] because the surface of this solid electrolyte was highly sensitive and thus reacted with a small amount of moisture and CO2 in the glove box. In contrast, the liquid-phase sample exhibited a peak at approximately 534 eV attributed to the O–C–O and C = O bonds present in alkyl carbonate lithium (ROCO2Li) [42], which was derived from the reaction between the solid-electrolyte surface and the ethyl propionate (CH3CH2COOC2H5). In addition, a new peak appeared at approximately 535 eV, which was attributed to the C–O–C bond [43]. The C 1 s spectrum of the mechanically milled sample exhibited a peak at approximately 288 eV, which was attributed to Li2CO3. In contrast, the C 1 s profile of the liquid-phase synthesized sample exhibited a significantly strong peak at approximately 286 eV, which was ascribed to the C–O–C bond [43], and a weak peak at approximately 288 eV, which corresponded to O–C–O and C = O [42]. These results show that the particle surfaces of the Li7P2S8I solid electrolyte synthesized by liquid-phase shaking were homogeneously covered by an organic-solvent-derived surface layer during the synthesis. In addition, solid-electrolyte components (S 2p) were detected in the wide-scan spectrum; therefore, the thickness of the surface layer in the liquid-phase synthesized sample was expected to be less than the escape depth of photoelectrons (several nanometers). Notably, although surface layers were detected in both the liquid-phase and mechanically milled samples, the difference in the homogeneity and chemical composition of these surface layers affected the lithium stability of the two types of samples, with the liquid-phase sample showing an electrochemically Li-stable surface layer.

XPS profiles (O 1s, C 1s, and S 2p) of Li7P2S8I solid electrolytes synthesized by mechanical milling and liquid-phase shaking
PDF analysis
Figure 5 shows the PDF analysis results for the Li7P2S8I solid electrolytes synthesized by mechanical milling and liquid-phase shaking. The glass structures of both samples comprised a P–S correlation within the PS4 molecule at a distance of 2.0 Å, an S–S correlation within the same molecule at 3.4 Å, and a peak at approximately 4.0 Å originating from an S–S correlation between the molecules [44, 45]. The difference in peak intensity can be attributed to compositional differences; however, there was no notable distinction in the P–S correlation or presence of PS4 molecules in each glass structure. This result indicated that the liquid-phase shaking sample exhibited only a PS4 tetrahedral structure, which was similar to that observed in a previous study [46]. However, the spectra for both samples differed above 5 Å, indicating that they comprised slightly different glass structures; a detailed analysis of the glass structures will be conducted in the future. Notably, a new peak at a bond length of approximately 1.6 Å was observed only in the sample synthesized by liquid-phase shaking. The spectrum of the ethyl propionate solvent was multiplied by a factor of 0.15 and the difference was calculated; as a result, the peak at 1.6 Å was much weaker and the peak at 2.7 Å disappeared (Fig. S4). This result indicated that the state of the ethyl propionate solvent remaining on the surface was different from pure ethyl propionate, suggesting that ethyl propionate chemically bonded with the particle surfaces of Li7P2S8I. These results are consistent with those obtained from the O 1 s and C 1 s XPS profiles.

PDF analysis results of Li7P2S8I solid electrolytes synthesized by mechanical milling and liquid-phase shaking
Neutron diffraction analysis
Figure 6 shows the neutron diffraction patterns and PDFs of the Li7P2S8I solid electrolytes synthesized by liquid-phase shaking and mechanical milling. The neutron diffraction pattern of the former exhibited a higher background than that of the latter. In addition, a tendency for the intensity to decrease with decreasing lattice spacing d was observed only for the Li7P2S8I solid electrolyte synthesized by liquid-phase shaking, indicating that it included H+ derived from the solvents.

Neutron diffraction pattens (a) and PDF of Li7P2S8I solid electrolytes (b) synthesized by liquid-phase shaking and mechanical milling
Thus, we could conclude that the solvent-derived organic surface layer formed on the Li7P2S8I solid-electrolyte particles acted as a solid-electrolyte interface (SEI) for the Li anode. To date, numerous studies on SEIs in organic-electrolyte batteries have been reported. In organic-electrolyte batteries, signals corresponding to the carbonyl group (C = O), polyether carbon (C–O–C), hydrocarbon (C–C/C–H), Li–F, LixPOyFz, and CF2 are generated at the Li metal anode owing to the decomposition of the carbonate solvents and LiPF6 [47,48,49], which stabilize the Li metal surface. Similarly, in this study, signals corresponding to the carbonyl group (C = O), polyether carbon (C–O–C), and hydrocarbon (C–C/C–H) were detected for the Li7P2S8I solid electrolyte surface synthesized by liquid-phase shaking. This surface layer is one of the factors that contributes to the high Li-metal stability in the Li7P2S8I solid electrolyte, synthesized using the liquid-phase shaking method, than in the mechanical milled electrolyte. In addition, the particle surfaces form grain boundaries during the fabricating of the molding body, and these grain boundaries may significantly influence the stability of Li metal. However, the contributions of the following three factors to the stability of Li metal cannot be excluded: (1) Primarily, the solvent-derived components in the solid-electrolyte particle may affect the stability of Li metal. Previous studies show that oxygen in the bulk also contributes to Li stability improvement [50, 51]. (2) The particle size also affects the stability of Li metal. In this study, the particle sizes of the liquid-phase shaking and mechanically milled samples were approximately 1 and 5 μm, respectively [26, 14]. A previous study suggests that small and uniform solid-electrolyte particles are less prone to physical degradation owing to the volume changes of the In–Li anode [52]. (3) The liquid-phase shaking sample includes LiI residue, which possesses superior electrochemical stability and prevents Li metal oxidation [53, 54]. Moreover, the surface layer, derived from the ethyl propionate solvent, and the LiI layer at the interface exhibit a synergistic effect.
Conclusion
Li7P2S8I solid electrolytes synthesized by liquid-phase shaking and mechanical milling were analyzed in terms of their local structures and particle surface states. Sample synthesized by both methods exhibited similar local structures: the main amorphous regions of both samples comprised nanocrystals such as LiI, Li4PS4I, and Li10P3S12I. These results indicated that crystalline components alone did not form ionic conductive pathways and that both the amorphous region and amorphous/crystalline interface contributed to the high ionic conductivity. In addition, DC polarization testing revealed that the Li7P2S8I synthesized by liquid-phase shaking was more stable toward Li metal than that synthesized by mechanical milling. Analyzing the particle surface states revealed that the particle surfaces of the sample synthesized by liquid-phase shaking included O–C–O–O and C = O bonds derived from ROCO2Li and a C–O–C bond with protons derived from the ethyl propionate solvent, which were not present for the mechanically milled sample. These surface layers were one of the factors that contributed to the high Li stability, a behavior similar to that observed in organic-electrolyte batteries. This study clarifies, for the first time, the glass-ceramic structure, which affects ionic conductivity, and the surface state of sulfide solid electrolytes, which contributes to stability toward Li. We expect this knowledge to contribute to the development of sulfide solid electrolytes by enabling efficient harnessing of the surface states of the particles.
References
Tufail MK, Zhai P, Jia M, Zhao N, Guo X (2023) Design of solid electrolytes with fast ion transport: computation-driven and practical approaches. Energy Mater Adv 4:0015. https://doi.org/10.34133/energymatadv.0015
Liu Z, Fu W, Payzant EA, Yu X, Wu Z, Dudney NJ, Kiggans J, Hong K, Rondinone AJ, Liang C (2013) Anomalous high ionic conductivity of nanoporous β-Li3PS4. J Am Chem Soc 135(3):975–978. https://doi.org/10.1021/ja3110895
Boulineau S, Courty M, Tarascon JM, Viallet V (2012) Mechanochemical synthesis of Li-argyrodite Li6PS5X (X = cl, br, I) as sulfur-based solid electrolytes for all solid-state batteries application. Solid State Ion 221:1–5. https://doi.org/10.1016/j.ssi.2012.06.008
Yamane H, Shibata M, Shimane Y, Junke T, Seino Y, Adams S, Minami K, Hayashi A, Tatsumisago M (2007) Crystal structure of a superionic conductor, Li7P3S11. Solid State Ion 178(15–18):1163–1167. https://doi.org/10.1016/j.ssi.2007.05.020
Kamaya N, Homma K, Yamakawa Y, Hirayama M, Kanno R, Yonemura M, Kamiyama T, Kato Y, Hama S, Kawamoto K, Mitsui A (2011) A lithium superionic conductor. Nat Mater 10(9):682–686. https://doi.org/10.1038/nmat3066
Kato Y, Hori S, Saito T, Suzuki K, Hirayama M, Mitsui A, Yonemura M, Iba H, Kanno R (2016) High-power all-solid-state batteries using sulfide superionic conductors. Nat Energy 1(4):1–7. https://doi.org/10.1038/nenergy.2016.30
Li Y, Song S, Kim H, Nomoto K, Kim H, Sun X, Hori S, Suzuki K, Matsui N, Hirayama M, Mizoguchi T (2023) A lithium superionic conductor for millimeter-thick battery electrode. Science 381(6653):50–53. https://doi.org/10.1126/science.add7138
Sakuda A, Hayashi A, Tatsumisago M (2013) Sulfide solid electrolyte with favorable mechanical property for all-solid-state lithium battery. Sci Rep 3(1):2261. https://doi.org/10.1038/srep02261
Hikima K, Totani M, Obokata S, Muto H, Matsuda A (2022) Mechanical properties of sulfide-type solid electrolytes analyzed by indentation methods. ACS Appl Energy Mater 5(2):2349–2355. https://doi.org/10.1021/acsaem.1c03829
Yang Q, Li C (2018) Li metal batteries and solid-state batteries benefiting from halogen-based strategies. Energy Storage Mater 14:100–117. https://doi.org/10.1016/j.ensm.2018.02.017
He B, Zhang F, Xin Y, Xu C, Hu X, Wu X, Yang Y, Tian H (2023) Halogen chemistry of solid electrolytes in all-solid-state batteries. Nat Rev Chem 7(12):826–842. https://doi.org/10.1038/s41570-023-00541-7
Zhang K, Jin Z (2022) Halogen-enabled rechargeable batteries: current advances and future perspectives. Energy Storage Mater 45:332–369. https://doi.org/10.1016/j.ensm.2021.11.048
Luo Q, Ming L, Zhang D, Wei C, Wu Z, Jiang Z, Liu C, Liu S, Cao K, Zhang L, Yu C (2023) Constructing Br-doped Li10SnP2S12-based all-solid-state batteries with superior performances. Energy Mater Adv 4:0065. https://doi.org/10.34133/energymatadv.0065
Choi SJ, Choi SH, Bui AD, Lee YJ, Lee SM, Shin HC, Ha YC (2018) LiI-doped sulfide solid electrolyte: enabling a high-capacity slurry-cast electrode by low-temperature post-sintering for practical all-solid-state lithium batteries. ACS Appl Mater Interfaces 10(37):31404–31412. https://doi.org/10.1021/acsami.8b11244
Spannenberger S, Miß V, Klotz E, Kettner J, Cronau M, Ramanayagam A, di Capua F, Elsayed M, Krause-Rehberg R, Vogel M, Roling B (2019) Annealing-induced vacancy formation enables extraordinarily high Li + ion conductivity in the amorphous electrolyte 0.33 LiI + 0.67 Li3PS4. Solid State Ion 341:115040. https://doi.org/10.1016/j.ssi.2019.115040
Song S, Hori S, Li Y, Suzuki K, Matsui N, Hirayama M, Saito T, Kamiyama T, Kanno R (2022) Material search for a Li10GeP2S12-type solid electrolyte in the Li–P–S–X (X = br, I) system via clarification of the composition–structure–property relationships. Chem Mater 34(18):8237–8247. https://doi.org/10.1021/acs.chemmater.2c01608
Wu Z, Wang R, Yu C, Wei C, Chen S, Liao C, Cheng S, Xie J (2022) Origin of the high conductivity of the LiI-doped Li3PS4 electrolytes for all-solid-state lithium–sulfur batteries working in wide temperature ranges. Ind Eng Chem Res 62(1):96–104. https://doi.org/10.1021/acs.iecr.2c04158
Wu Z, Chen S, Yu C, Wei C, Peng L, Wang HL, Cheng S, Xie J (2022) Engineering high conductive Li7P2S8I via Cl-doping for all-solid-state Li-S batteries workable at different operating temperatures. Chem Eng J 442:136346. https://doi.org/10.1016/j.cej.2022.136346
Rangasamy E, Liu Z, Gobet M, Pilar K, Sahu G, Zhou W, Wu H, Greenbaum S, Liang C (2015) An iodide-based Li7P2S8I superionic conductor. J Am Chem Soc 137(4):1384–1387. https://doi.org/10.1021/ja508723m
Hikima K, Yamamoto T, Phuc NH, Matsuda R, Muto H, Matsuda A (2020) Improved ionic conductivity of Li2S-P2S5-LiI solid electrolytes synthesized by liquid-phase synthesis. Solid State Ion 354:115403. https://doi.org/10.1016/j.ssi.2020.115403
Ujiie S, Hayashi A, Tatsumisago M (2013) Preparation and ionic conductivity of (100 – x)(0.8Li2S·0.2P2S5)·xLiI glass–ceramic electrolytes. J Solid State Electrochem 17:675–680. https://doi.org/10.1007/s10008-012-1900-7
Ujiie S, Hayashi A, Tatsumisago M (2012) Structure, ionic conductivity and electrochemical stability of Li2S–P2S5–LiI glass and glass–ceramic electrolytes. Solid State Ion 211:42–45. https://doi.org/10.1016/j.ssi.2012.01.017
Lu S, Kosaka F, Shiotani S, Tsukasaki H, Mori S, Otomo J (2021) Optimization of lithium ion conductivity of Li2S-P2S5 glass ceramics by microstructural control of crystallization kinetics. Solid State Ion 362:115583. https://doi.org/10.1016/j.ssi.2021.115583
Tsukasaki H, Mori S, Morimoto H, Hayashi A, Tatsumisago M (2017) Direct observation of a non-crystalline state of Li2S–P2S5 solid electrolytes. Sci Rep 7(1):4142. https://doi.org/10.1038/s41598-017-04030-y
Phuc NH, Morikawa K, Mitsuhiro T, Muto H, Matsuda A (2017) Synthesis of plate-like Li3PS4 solid electrolyte via liquid-phase shaking for all-solid-state lithium batteries. Ionics 23:2061–2167. https://doi.org/10.1007/s11581-017-2035-8
Hikima K, Ogawa K, Muto H, Matsuda A (2022) Li7P2S8I solid electrolytes synthesized by liquid-phase synthesis with improved heat treatment process. J Ceram Soc Jpn 130(4):299–302. https://doi.org/10.2109/jcersj2.21176
Nishibori E, Takata M, Kato K, Sakata M, Kubota Y, Aoyagi S, Kuroiwa Y, Yamakata M, Ikeda N (2001) The large debye–scherrer camera installed at SPring-8 BL02B2 for charge density studies. J Phys Chem Solids 62(12):2095–2098. https://doi.org/10.1016/S0168-9002(01)00639-8
Tsukasaki H, Otoyama M, Kimura T, Mori S, Sakuda A, Hayashi A, Tatsumisago M (2020) Exothermal behavior and microstructure of a LiNi1/3Mn1/3Co1/3O2 electrode layer using a Li4SnS4 solid electrolyte. J Power Sources 479:228827. https://doi.org/10.1016/j.jpowsour.2020.228827
Yao B, Sun T, Warren A, Heinrich H, Barmak K, Coffey KR (2010) High contrast hollow-cone dark field transmission electron microscopy for nanocrystalline grain size quantification. Micron 41(3):177–182. https://doi.org/10.1016/j.micron.2009.11.008
Kulovits AK, Facco G, Wiezorek JM (2012) Grain size determination in nano-scale polycrystalline aggregates by precession illumination-hollow cone dark field imaging in the transmission electron microscope. Mater Charact 63:17–26. https://doi.org/10.1016/j.matchar.2011.10.003
Lábár JL (2005) Consistent indexing of a (set of) single crystal SAED pattern(s) with the process diffraction program. Ultramicroscopy 103(3):237–249. https://doi.org/10.1016/j.ultramic.2004.12.004
Lábár JL (2008) Electron diffraction based analysis of phase fractions and texture in nanocrystalline thin films, part I: principles. Microsc Microanal 14(4):287–295. https://doi.org/10.1017/S1431927608080380. Scopus
Yamada H, Nakada K, Takemoto M, Ohara K (2022) Fully automated measurement system for temperature-dependent X-ray total scattering at beamline BL04B2 at SPring-8. J Synchrotron Radiat 29(2):549–554. https://doi.org/10.1107/S1600577521013527. From NLM Medline
Ohara K, Onodera Y, Murakami M, Kohara S (2021) Structure of disordered materials under ambient to extreme conditions revealed by synchrotron X-ray diffraction techniques at SPring-8—Recent instrumentation and synergic collaboration with modelling and topological analyses. J Phys Condens Matter 33(38):383001. https://doi.org/10.1088/1361-648X/ac0193
Sedlmaier SJ, Indris S, Dietrich C, Yavuz M, Dräger C, von Seggern F, Sommer H, Janek J (2017) Li4PS4I: a Li + superionic conductor synthesized by a solvent-based soft chemistry approach. Chem Mater 29(4):1830–1835. https://doi.org/10.1021/acs.chemmater.7b00013
Yamamoto T, Phuc NH, Muto H, Matsuda A (2019) Preparation of Li7P2S8I solid electrolyte and its application in all-solid-state lithium-ion batteries with graphite anode. Electron Mater Lett 15:409–414. https://doi.org/10.1007/s13391-019-00133-y
Choi SJ, Lee SH, Ha YC, Yu JH, Doh CH, Lee Y, Park JW, Lee SM, Shin HC (2018) Synthesis and electrochemical characterization of a glass-ceramic Li7P2S8I solid electrolyte for all-solid-state Li-ion batteries. J Electrochem Soc 165(5):A957. https://doi.org/10.1149/2.0981805jes
Mercier R, Malugani JP, Fahys B, Robert G (1981) Superionic conduction in Li2S-P2S5-LiI-glasses. Solid State Ion 5:663–666. https://doi.org/10.1016/0167-2738(81)90341-6
Hayashi A, Noi K, Sakuda A, Tatsumisago M (2012) Superionic glass-ceramic electrolytes for room-temperature rechargeable sodium batteries. Nat Commun 3(1):856. https://doi.org/10.1038/ncomms1843
Ikeda K, Kimura T, Ohara K, Sato T, Ohshita H, Sakuda A, Hayashi A (2023) Vacancies introduced during the crystallization process of the glass-ceramics superionic conductor, Na3PS4, investigated by neutron total scattering and reverse Monte Carlo method. J Phys Chem C 127(13):6199–6206. https://doi.org/10.1021/acs.jpcc.3c00414
Philippe B, Hahlin M, Edström K, Gustafsson T, Siegbahn H, Rensmo H (2015) Photoelectron spectroscopy for lithium battery interface studies. J Electrochem Soc 163(2):A178. https://doi.org/10.1149/2.0051602jes
Ding F, Xu W, Chen X, Zhang J, Engelhard MH, Zhang Y, Johnson BR, Crum JV, Blake TA, Liu X, Zhang JG (2013) Effects of carbonate solvents and lithium salts on morphology and coulombic efficiency of lithium electrode. J Electrochem Soc 160(10):A1894. https://doi.org/10.1149/2.100310jes
Ding D, Maeyoshi Y, Kubota M, Wakasugi J, Kanamura K, Abe H (2021) Non-flammable super-concentrated polymer electrolyte with solvated ionic liquid for lithium-ion batteries. J Power Sources 506:230099. https://doi.org/10.1016/j.jpowsour.2021.230099
Ohara K, Mitsui A, Mori M, Onodera Y, Shiotani S, Koyama Y, Orikasa Y, Murakami M, Shimoda K, Mori K, Fukunaga T (2016) Structural and electronic features of binary Li2S-P2S5 glasses. Sci Rep 6(1):21302. https://doi.org/10.1038/srep21302
Yamaguchi H, Kobayashi K, Hiroi S, Utsuno F, Ohara K (2023) Structural analysis and ionic conduction mechanism of sulfide-based solid electrolytes doped with br. Sci Rep 13(1):16063. https://doi.org/10.1038/s41598-023-43347-9
Indrawan RF, Yamamoto T, Nguyen HH, Muto H, Matsuda A (2020) Liquid-phase synthesis of 100Li3PS4-50LiI-xLi3PO4 solid electrolytes. Solid State Ion 345:115184. https://doi.org/10.1016/j.ssi.2019.115184
Bae J, Qian Y, Li Y, Zhou X, Goodenough JB, Yu G (2019) Polar polymer–solvent interaction derived favorable interphase for stable lithium metal batteries. Energy Environ Sci 12(11):3319–3327. https://doi.org/10.1039/C9EE02558H
Ma Q, Zhang X, Wang A, Xia Y, Liu X, Luo J (2020) Stabilizing solid electrolyte interphases on both anode and cathode for high areal capacity, high-voltage lithium metal batteries with high Li utilization and lean electrolyte. Adv Funct Mater 30(32):2002824. https://doi.org/10.1002/adfm.202002824
Li X, Zheng J, Ren X, Engelhard MH, Zhao W, Li Q, Zhang JG, Xu W (2018) Dendrite-free and performance-enhanced lithium metal batteries through optimizing solvent compositions and adding combinational additives. Adv Energy Mater 8(15):1703022. https://doi.org/10.1002/aenm.201703022
Indrawan RF, Matsuda R, Hikima K, Matsuda A (2023) Enhanced electrochemistry stability of oxygen doped Li6PS5Cl argyrodite solid electrolyte by liquid-phase synthesis. Solid State Ion 401:116344. https://doi.org/10.1016/j.ssi.2023.116344
Wu M, Liu G, Yao X (2022) Oxygen doped argyrodite electrolyte for all-solid-state lithium batteries. Appld Phys Lett 121(20):203904. https://doi.org/10.1063/5.0114275
Kim HS, Watanabe K, Matsui N, Suzuki K, Kanno R, Hirayama M (2023) Crack suppression by downsizing sulfide-electrolyte particles for high-current-density operation of metal/alloy anodes. Batter Supercaps 6(10):e202300306. https://doi.org/10.1002/batt.202300306
Yang S, Yamamoto K, Mei X, Sakuda A, Uchiyama T, Watanabe T, Takami T, Hayashi A, Tatsumisago M, Uchimoto Y (2021) High rate capability from a graphite anode through surface modification with lithium iodide for all-solid-state batteries. ACS Appl Energy Mater 5(1):667–673. https://doi.org/10.1021/acsaem.1c03166
Wang Y, Liang J, Song X, Jin Z (2023) Recent progress in constructing halogenated interfaces for highly stable lithium metal anodes. Energy Storage Mater 54:732–775. https://doi.org/10.1016/j.ensm.2022.10.054
Acknowledgements
The XPS experiments were conducted at BL7U at the Aichi Synchrotron Radiation Center, Aichi Science & Technology Foundation, Aichi, Japan (Proposal No. 202204118 and No. 202302109). The synchrotron radiation experiments were performed with the approval of the Japan Synchrotron Radiation Research Institute (Proposal No. 2023A1001, No. 2022B1833). The neutron total scattering experiments were approved by the Neutron Scattering Program Advisory Committee (Proposal No. 2019S06). This work was partially supported by MEXT Promotion of Distinctive Joint Usage/Research Center Support Program Grant Number JPMXP0723833161. We thank Ms. Masayo Takahashi and Ms. Reiko Matsuda for their assistance with the experiments.
Funding
This work was supported by JSPS KAKENHI (Grant Nos. JP 19H05814, JP 22H04614, JP22H04616), the Advanced Low Carbon Technology Specially Promoted Research for Innovative Next-Generation Batteries (JST-ALCA-SPRING, JPMJAL1301) program of the Japan Science and Technology Agency (JST), and the SOLiD-EV project (JPNP18003) of the New Energy and Industrial Technology Development Organization (NEDO), Japan.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hikima, K., Ogawa, K., Indrawan, R.F. et al. Structure and particle surface analysis of Li2S–P2S5–LiI-type solid electrolytes synthesized by liquid-phase shaking. J Solid State Electrochem (2024). https://doi.org/10.1007/s10008-024-05898-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10008-024-05898-3