
Abstract
Various transition metal dichalcogenides materials have been investigated from bulk to monolayer phases for different advanced technological applications. Tin disulfide monolayer offers advantages as an anode material for Li/Na-ion batteries, although it cannot be considered ideal for direct exploitation. We systematically performed a comparative study of the adsorption and diffusion behaviour of Li/Na on a pristine SnS\(_2\) monolayer and on a SnS\(_2\) monolayer with S-vacancy for enhancement of electrochemical performance, using density functional theory approach. Although all the adsorption sites are exothermic, it was established that Li/Na adatoms mostly prefer to bind strongly on SnS\(_2\) monolayer with S-vacancy but avoiding the S-vacancy site. It was established that avoiding the S-vacancy site along the path, excellent diffusion barriers of 0.19 eV for Li and 0.13 eV for Na were achieved, suggesting possible ultrafast charge/discharge rate. Due to reduced molar mass, the SnS\(_2\) monolayer with S-vacancy has a slightly higher storage capacity than its pristine counterparts for both Li and Na adatoms. The obtained open circuit voltage values are within the range of 0.25–3.00 V assuring that the formation of dendrites can surely be averted for the envisaged battery operation. Understanding the effects of an S-vacancy on the electrochemical properties of Li/Na on the SnS\(_2\) monolayer allows us to consider possible improvements to energy storage devices that can be applied as a result of improved anode material.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lithium and sodium represent a large fraction of the materials used in high density energy storage [1]. The continuous rising global demand of Li-ion batteries (LIBs) will lead to relative scarcity of lithium material, an exploration of other similar materials is a matter of urgency [2]. In comparison, the sodium of Na-ion batteries (NIBs), while having similar properties to Li, is more abundant and cheaper [3, 4]. Just like the Li ions in LIBs, the Na ions in NIBs require a suitable platform material acting as electrode specifically anode for good performance, with a graphite material serving a purpose due to its abundance and non-toxicity [5]. The improvement of performance of both LIBs and NIBs using electrode materials other than graphite (low storage capacity and poor rate capability) is always necessary [6]. Astonishing physical properties of two-dimensional (2D) materials such as the excellent electrical conductivity, high mechanical strength, low dimensionality, excellent dynamic stability and energetic stability [7,8,9,10,11] encourage researchers for exploration of energy harvesting and storage in LIBs and NIBs, an important field for future energy security, affordability and efficiency.
Graphene [12] has been researched in depth and has proven to have exciting peculiar properties warranting it to act as anode material for LIBs [13, 14], its setback is due to weaknesses of Li binding, leading to the formation of clusters that can nucleate to undesired dendrites [15,16,17,18,19]. Thus, the theoretical and experimental avenues of improving binding in graphene were explored [20, 21]. Chemical modification (vacancies and atomic doping) to create reactive sites for Li adatoms brought forward promising results for improving novelty of graphene for energy application [21,22,23,24,25,26]. Apart from graphene and chemically modified graphene, the focus is further directed to other various 2D transition metal dichalcogenides (TMDs) [27,28,29,30,31,32,33,34] which have attracted immense attention of researchers due to their promise of serving as the electrode materials. The most popular explored TMDs is the hexagonal 2D tin disulfide (SnS\(_2\)), which consists of three trigonal subplanes sandwiching an Sn-subplane between two S-subplanes.
SnS\(_2\) monolayer exists in nature as various polytypes made up of H or T phases. The 1H-SnS\(_2\) phase belongs to the \(P\bar{6}m2\) group with ABA stacked subplanes, and the 1T-SnS\(_2\) phase belongs to the \(P\bar{3}m2\) group with ABC stacked subplanes [34, 35]. The H and T phases are used to denote some polytypes of multi-layer SnS\(_2\), designating the number of layers and their phase types, such as 1T, 2H, and 4H, with an additional polytype being denoted as 18R. When comparing the 1T-SnS\(_2\) and 1H-SnS\(_2\) monolayers, the former has been found to be energetically more stable [36, 37]. Since material stability is an important consideration for implementation in nanotechnology, 1T-SnS\(_2\) can be the preferred polytype for anode materials.
SnS\(_2\) monolayer possesses a larger lattice cell size leading to high surface area in comparison to graphene [38], resulting in a higher storage capacity for the adsorption of Li/Na [36, 39]. Wang et al. used solvothermal route to synthesise anodes of 2D SnS\(_2\) that have a capacity of 1200 mAh/g for LiBs and 600 mAh/g for NIBs [40]. Theoretically, SnS\(_2\) monolayer has a high capacity of 1231 mAh/g for LIBs and 1136 mAh/g for NIBs [41]. The large storage capacity is a requirement for accommodating as many ions as possible for use in batteries. The SnS\(_2\) monolayer has poor conductivity due to its large band gap and a relatively large diffusion barrier possibly leading to a negative impact on a charging/discharging cycling performance [41].
For SnS\(_2\) monolayer to be a high-performance energy electrode material, a high adatom binding energy (high stability), high number of reactive sites to improve storage capacity, more pronounced electronic states within the band gap (high electronic conductivity), etc. are required [36, 42, 43]. High-performance energy electrode material will make it possible for high energy demand applications and predicting new energy storage technologies. The effort to improve the energy density, battery cycle life, and power density in the SnS\(_2\) monolayer has been an on going work. More recently, previous studies have been undertaken to study the SnS\(_2\) multilayers interactions for LIBs [44]. The studies of SnS\(_2\) monolayer coupled with graphene-like materials for enhancing electronic conductivity and mechanical strength [35, 45, 46] were considered unabated. The influence of atomic doping in SnS\(_2\) monolayer has also been tested with respect to energetic stability and electrochemical properties for energy storage [41, 47,48,49].
The introduction of an S-vacancy in the SnS\(_2\) monolayer, particularly as an anode material has experimentally been explored and verified with DFT calculations. Zhang et al. [50] synthesised SnS\(_2\) nanoplates with abundant S-vacancies using facile solvothermal techniques. Material characterisation and electrochemical measurements analysis revealed that SnS\(_2\) nanoplates with S-vacancies have a small band gap leading to a better electronic conductivity, high Li-ion diffusion mobility, enhanced cycling performance and high rate capability. Huang et al. [51] synthesised what is called binder-free SnS\(_2\) sheet array with high S-vacancy concentration using hydrothermal technique for LIBs exploitation. It was reported that this composite binder-free SnS\(_2\) sheet array with high S-vacancy concentration has a high capacity of 2073.8 mAh/g, whereas the pristine SnS\(_2\) powders yields only 400.3 mAh/g [51]. DFT calculations indicated that SnS\(_2\) monolayer with high S-vacancy concentration has lower band gap of 0.517 eV, suggesting an enhanced electronic conductivity suitable for LIBs [51]. Wang et al. synthesised SnS\(_2\) microspheres with S-vacancies through a one-step solvothermal process, and the excellent high capacity of 486.2 mAh/g was achieved for NIB applications [52]. DFT calculations of SnS\(_2\) microsphere with S-vacancies revealed a decreased band gap suggesting a good conductivity [52]. There is enough evidence of good electrochemical similarity between LIBs and SIBs more especially when SnS\(_2\) monolayer is employed as an anode. The basic information of comparison between Li and Na (differing in atomic radii) on SnS\(_2\) monolayer with S-vacancy is not sufficient more especially on their stability, diffusion mobility etc. using DFT methods.
In this work, we performed DFT calculations to systematically compare the energetic stability and electrochemical properties of the adsorption of Li/Na on a pristine SnS\(_2\) monolayer and SnS\(_2\) monolayer with S-vacancy structures. We look for improvements in the Li/Na binding energies, voltages, storage capacities and electronic conductivity (density of states) through Li/Na adsorption on a SnS\(_2\) monolayer with S-vacancy. Our results indicate that Li/Na adatoms prefer to bind strongly on SnS\(_2\) monolayer with S-vacancy but avoiding the S-vacancy site. We also considered the diffusion of Li/Na across the SnS\(_2\) monolayer with S-vacancy by calculating activation energies, and it was established that avoiding the S-vacancy site along the path, an improved diffusion barrier of 0.19 eV for Li and 0.13 eV for Na is achieved. The open circuit voltage average for Li (Na) on a pristine SnS\(_2\) monolayer is 2.06 V (2.03 V) and that on SnS\(_2\) monolayer with S-vacancy is 2.16 V (2.24 V). As a result, these material systems will be able to yield significant charging voltage and thus, opening the door to experimental studies into the viability of S-vacancies or vacancies in general in similar 2D materials.
Following this introduction (“Introduction”), we summarise the computational methodology used to perform our calculations and obtain results (“Methodology”), list and discuss these results and what can be derived from them with respect to the properties or SnS\(_2\) (“Results and discussion”), and summarise our overall results and what they mean for further studies regarding SnS\(_2\) and similar 2D materials (“Conclusion”).
Methodology
Structural optimizations and calculations of various properties of Li/Na on the SnS\(_2\) monolayer were carried out using the first-principles density functional theory (DFT) methods [53]. The plane wave based DFT methods with the projected augmented wave (PAW) method for pseudopotential generation [54] are implemented within the Quantum ESPRESSO software suite [55]. Before each calculation can begin, the electronic spin is imposed on each atom. For the description of the electron exchange correlation potential, the generalised gradient approximation with the Perdew-Burke-Ernzerhof (GGA-PBE) flavour was adopted [56]. The kinetic energy cutoff for expansion of the plane wave was set to 45 Ry (\(\sim\)600 eV), while that of charge densities was set to 300 Ry (\(\sim\)4000 eV). These kinetic energy cutoff values were chosen after convergence tests. Total energies and forces convergence criteria were set to differences less than 1\(\times\)10\(^{-4}\) eV and 1\(\times\)10\(^{-3}\) eV/Å, respectively.
A Monkhorst-Pack (MP) scheme [57] with a k-mesh of 5\(\times\)5\(\times\)1 sufficient to give a converged total energy was used for the generation of the k-points for sampling the Brillouin zone. Additionally, the electronic occupancy was set using the Gaussian smearing method [58], with a spread of 0.01 Ry, to simplify the calculation at the occupied energy states.
For the calculation of density of states (DOS), we aimed for greater accuracy of the energy levels, since the GGA-PBE calculation alone tends to give underestimated energy levels. To correct this error, DFT plus U with a Hubbard-U correction parameter of 8 was used after a series of tests was performed. Furthermore, it was found that this U value mainly improves the highest energy bands of S atom. This correction allows for an estimation of band gap closer to experimental values. Additionally, the k-mesh is doubled to 10\(\times\)10\(\times\)1, for the correct exploitation of hubbard-U parameter. The Tetrahedra method [59] was employed for electron occupancy.
To ensure the isolation of single Li on the SnS\(_2\) monolayer supercell, the periodic boundary conditions are obeyed. The SnS\(_2\) supercell test of convergence was performed starting with 2\(\times\)2, 3\(\times\)3, 4\(\times\)4, 5\(\times\)5 and 6\(\times\)6 sizes containing one Li adatom. The Li binding energy differences for these supercells are 0.199 eV (2\(\times\)2 and 3\(\times\)3), 0.018 eV (3\(\times\)3 and 4\(\times\)4), 0.013 (4\(\times\)4 and 5\(\times\)5) and 0.010 (5\(\times\)5 and 6\(\times\)6). Although the test results start to converge at 3\(\times\)3, the larger 5\(\times\)5 supercell has been adopted for all configurations studied. To avoid spurious multi-layer interactions, the vacuum space was set to a converged 15 Å between SnS\(_2\) layers. To take into account the influence of the van der Waals dispersion corrections on the energetic and structural stability, the Grimme dispersion correction method (DFT-D3) was adopted [60].
To investigate Li/Na diffusion across the SnS\(_2\) monolayer, the nudged elastic band (NEB) method [61] was employed. This was used to obtain a profile of the minimum energy path between different sites of interest, whereby the diffusion barrier can be calculated.
Results and discussion
Structures considered
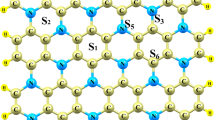
Having disregarded the less stable 1H phase of SnS\(_2\), we consider the 5\(\times\)5 supercell constructed using a thermodynamic stable 1T phase shown in Fig. 1. The chosen supercell size was selected for having a greater surface area to allow the creation of the S-vacancy at the middle of the cell obeying principle of boundary conditions, and also allowing us to investigate the different Li/Na adatom sites closer or further away from the S-vacancy. In this study, Li/Na adatom configurations on the different sites namely H\(_{SnS_2}\), T\(_{Sn}\), T\(_S\), and V\(_S\), representing the position on top of a lower layer S (Hexagon site), on top of a central layer Sn, on top of an upper layer S, and on top of the S-vacancy site are examined, respectively. These unique adatom sites were identified focusing on the lattice symmetry of SnS\(_2\) monolayer as shown in Fig. 1.

A 5\(\times\)5\(\times\)1 supercell of SnS\(_2\) with adatom sites of interest. The sites are named T\(_S\), H\(_{SnS_{2}}\), T\(_{Sn}\), and V\(_S\) representing on top S atom, at the centre of hexagon, on top of Sn atom and at the S-vacancy, respectively
In the next section, the results of energetic stability and relaxed structures of Li/Na adatom configurations on H\(_{SnS_2}\), T\(_{Sn}\) and T\(_S\) on a pristine SnS\(_2\) monolayer are compared with those next to the S-vacancy (H\(_{SnS_2}^*\) T\(_{Sn}^*\) and T\(_{S}^*\) sites).
Energetic stability and structural properties
To gain insight into the energetic stability of Li/Na adatom configurations at H\(_{SnS_2}\), T\(_{Sn}\), T\(_S\), and V\(_S\) sites, we calculated their binding energies E\(_{b}\) (measured in eV) as the energy difference between the relaxed adatom bonded with the SnS\(_2\) monolayer, isolated SnS\(_2\) monolayer and isolated Li/Na adatom, clearly described by the following expression;
with \(E_{SnS_2+ad}\) been the total energy of relaxed Li/Na adatom bonded with the SnS\(_2\) monolayer, \(E_{SnS_2}\) is the total energy of the SnS\(_2\) monolayer, \(E_{ad}\) is the energy of an isolated Li/Na adatom, and n is the number of adatoms adsorbed on the monolayer. In this work, the E\(_{b}\) of Li configurations as well as Na configurations at the different sites are indicated in Table 1 and their relaxed configurations are presented in the supplementary figure 1. E\(_{b}\) describes the energy needed to detach adatoms from the substrate, as such, positive value reveals that adatoms would be attracted to the SnS\(_2\) monolayer and form a uniform Li/Na adatom dispersion, while the negative value reveals a possible agglomeration of adatoms forming an undesired bulk metallic material above the substrate. Instinctively, as shown in Table 1, all the configurations have positive binding energy values (suggesting exothermic reaction), indicating that the SnS\(_2\) monolayer is a suitable platform for interacting with Li/Na adatoms. Since they are not showing any possibility of forming a metallic Li cluster after structural relaxation.
Comparing the H\(_{SnS_2}\), T\(_{Sn}\) and T\(_S\) sites on the pristine SnS\(_2\) monolayer, Table 1 shows that H\(_{SnS_2}\) and T\(_{Sn}\) are the most stable sites with almost equal E\(_{b}\). This suggests that the Li/Na dispersion on H\(_{SnS_2}\) and T\(_{Sn}\) could co-exist under conditions of finite temperature. This could be attributed to the fact that the Li/Na atoms interact with equal number of nearest neighbour S atoms in both sites (Fig. 4). T\(_S\) is the least stable site for both Li and Na adatom configurations. In this T\(_S\) site, an interaction of Li/Na with its nearest neighbour Sn atoms is less energetically favourable. Table 1 shows that our calculated E\(_{b}\) values for H\(_{SnS_2}\) and T\(_{Sn}\) configurations are comparable to those reported by Lv et al. [62], calculated from the same exchange correlation functional (GGA-PBE). The E\(_{b}\) results of Samad et al. [35] for Na on pristine SnS\(_2\) monolayer configurations are smaller by more than 1.0 eV when compared to our values (same GGA-PBE functionals used), the underestimation likely resulting from a used small energy cutoff of 350 eV.

Top and side views of charge density difference of Li/Na adatoms on SnS\(_2\) monolayer calculated considering the issovalue of 0.002 electrons/bohr\(^3\). The purple colour shows the region of charge density decrease while the cyan colour shows the charge density increase
The E\(_{b}\) values of Li/Na configurations on V\(_{S}\) site and on its surroundings T\(_{Sn}^*\), T\(_{S}^*\) and H\(_{SnS_2}^*\) sites are also shown in Table 1. It is noted that the E\(_{b}\) values for T\(_{Sn}^*\), T\(_{S}^*\) and H\(_{SnS_2}^*\) sites surrounding V\(_{S}\) are significantly more than those of their pristine counterparts. In this situation, the most stable sites are still T\(_{Sn}^*\) and H\(_{SnS_2}^*\), and the binding energy of the former has increased considerably. The Li/Na on T\(_{Sn}^*\) and H\(_{SnS_2}^*\) sites have the same 1\(^{st}\) nearest neighbour S atoms or the same coordination number. The V\(_{S}\) site is the least stable for both the Li and Na configurations, due to its 1\(^{st}\) nearest neighbour atoms being Sn with less electronegativity (See Supplementary Fig. 1). Various studies confirmed this observation and reported electronegativity as governing factor [51, 63,64,65]. The improved E\(_{b}\) values suggest that Li/Na would prefer to be dispersed above the T\(_{Sn}^*\) and H\(_{SnS_2}^*\) sites without forming a metallic cluster. Although the E\(_{b}\) results indicate that pristine SnS\(_2\) is ideally suitable for the LIBs as an anode material, the s-vacancy suggests a possibility of high-performance LIBs.
Table 1 shows the values of relaxed Li/Na heights (d\(_{Li/Na-SnS_2}\)) above the sites. The most stable site H\(_{SnS_2}\) (although T\(_{Sn}\) has a very similar binding energy) has a short d\(_{Li-SnS_2}\) of 1.42 Å and d\(_{Na-SnS_2}\) of 1.92 Å. The shortest binding lengths appear at T\(_{Sn}\). The least stable site T\(_S\) has the longest height in both the alkali metal adsorptions. Due to the difference in electronegativity between S and Sn atoms, the 1\(^{st}\) nearest neighbour S atoms in H\(_{SnS_2}\) are likely to accept the Li/Na valence electron forming a strong ionic bonding with higher binding energy (responsible for shorter d\(_{Li/Na-SnS_2}\)) than the 1\(^{st}\) nearest neighbour Sn atoms in T\(_S\). Figure 2 presents the electronic charge distribution around the Li/Na adatom above the H\(_{SnS_2}\) stable site of pristine SnS\(_2\) monolayer as well as the SnS\(_2\) monolayer with vacancy. From the figures it is notable that charge is concentrated nearby the adatoms, indicating that the Li/Na adatom greatly influences the uniform charge distribution of SnS\(_2\) monolayer. Furthermore, it was found that the charge mainly depletes (purple) from the Li/Na adatoms (pronounced) towards S atoms (few). The charge accumulates (cyan) at the middle of bond lengths between S and Li atoms.

The variation in binding energies with respect to the gradual increasing of Li/Na content x (from Li/Na\(_x\)SnS\(_2\)) adsorbed on H\(_{SnS_2}\) and H\(^*_{SnS_2}\) sites in the pristine and S-vacancy SnS\(_2\) monolayer
We further examine the effect of multiple Li/Na atoms on the stability, by calculating the E\(_{b}\) with respect to the gradual increasing of the number of adatoms, as shown in Fig. 3. The Li/Na atoms are mostly adsorbed on the most energetically stable site ( H\(_{SnS_2}\) and H\(^*_{SnS_2}\)) and are increased to match the number of Sn atoms on the supercell. The gentle curvature of the graphs (Fig. 3) is a predictor that SnS\(_2\) can undergo a greater degree of lithiation/sodiation without clustering, likely resulting in a high storage capacity. Figure 3 also shows that E\(_{b}\) decreases as the number of Li/Na increases. This reduction in E\(_{b}\) could be attributed to electrostatic repulsive force arising between Li/Na atoms. The E\(_{b}\) reduces to 2.57 eV (2.61 eV) for H\(_{SnS_2}\) in pristine SnS\(_2\) (H\(^*_{SnS_2}\) in SnS\(_2\) with S-vacancy) for 25 Li adatoms and reduces to 1.50 eV (1.79 eV) for T\(_{Sn}\) in pristine SnS\(_2\) (H\(^*_{SnS_2}\) in SnS\(_2\) with S-vacancy) for 25 Na atoms. This decrease in E\(_{b}\) is an expected result of an increased number of adatoms, similar to results from comparable studies on graphene-like materials [64,65,66,67]. The binding energies are still far too high to approach 0.00 eV suggesting that more adatoms can still be accommodated before metallic Li/Na forms.
Diffusion of Li/Na adatom on SnS\(_2\) monolayer
We examined the Li/Na mobility on pristine SnS\(_2\) monolayer and on S-vacancy in the SnS\(_2\) monolayer by calculating the diffusion barriers. As discussed in “Methodology”, we performed NEB calculations to obtain saddle points and minimum energy paths following the fixed sites shown in Fig. 1. The Li/Na adatom passes through four fixed points (sites) on the SnS\(_2\) monolayer along the path as follows; T\(_S\) site \(\rightarrow\) H\(_{SnS_2}\) site \(\rightarrow\) T\(_{Sn}\) site \(\rightarrow\) T\(_S\) site for pristine SnS\(_2\) monolayer and T\(_S\) site \(\rightarrow\) H\(_{SnS_2}\) site \(\rightarrow\) T\(_{Sn}\) site \(\rightarrow\) T\(_S\) for S-vacancy in the SnS\(_2\) monolayer. The resulting relative energies along the path were plotted in Fig. 4a, b for Li and Na, respectively.

Relative energies and diffusion barriers of an a Li or b Na adatom on SnS\(_2\) monolayer. The Li/Na adatom follows T\(_S\) site \(\rightarrow\) H\(_{SnS_2}\) site \(\rightarrow\) T\(_{Sn}\) site \(\rightarrow\) T\(_S\) site for pristine SnS\(_2\) monolayer and T\(_S\) site \(\rightarrow\) H\(_{SnS_2}\) site \(\rightarrow\) T\(_{Sn}\) site \(\rightarrow\) V\(_S\) for S-vacancy in the SnS\(_2\) monolayer
Focusing on the pristine SnS\(_2\) monolayer, the T\(_{Sn}\) and H\(_{SnS_2}\) sites are clearly visible as having a highest relative energy peaks (Fig. 4a, b), which correlate directly with their highest binding energies. The diffusion barriers or activation energies recorded at the highest peaks are 0.612 eV (Li) and 0.417 eV (Na) for pristine SnS\(_2\) monolayer. These results agree quite well with those of Li on SnS (0.45 eV) [68]. These results are also on the same range with other graphene-like materials, such as borophene (0.40 eV for Li, 0.22 eV for Na) [69], MoS\(_2\) (0.47 eV for Li, 0.11 eV for Na) [46, 70], MoO\(_2\) (0.13 eV for Na) [67]. As shown in Figs. 4a, b, the effect of V\(_S\) increases the relative energy peak at H\(_{SnS_2}\) to 0.83 eV (Li) and 0.43 eV (Na) and reduces at T\(_{Sn}\). It becomes apparent that the S-vacancy in SnS\(_2\) monolayer has high diffusion barrier but the V\(_S\) site itself has a zero relative energy. Our calculated diffusion barriers are within the category of suitable candidates for LiB and NiB. However, our diffusion barriers could be much improved if a more efficient path across the SnS\(_2\) monolayer was selected, avoiding the V\(_S\) site. Considering Fig. 4, a path limited to moving as H\(_{SnS_2}\) \(\rightarrow\) T\(_{Sn}\), which when looking at Fig. 1 still allows for movement to all H\(_{SnS_2}\) sites on the SnS\(_2\) monolayer, gives us a much improved diffusion barrier of \(\sim\)0.19 eV for Li and \(\sim\)0.13 eV for Na, a significant improvement suggesting much faster atom diffusion.

The spin-up (green) and spin-down (blue) total density of states for the a pristine and b S-vacancy SnS\(_2\) monolayers. The total density of states also depicts the following: Li on c pristine and d S-vacancy SnS\(_2\) monolayers as well as Na on e pristine and f S-vacancy SnS\(_2\) monolayers
Electronic properties
We further investigated the electronic conductivity of SnS\(_2\) monolayer using total density of states (TDOS), in order to understand its electrochemical performance. The TDOS studied are for the pristine SnS\(_2\) monolayer and S-vacancy SnS\(_2\) monolayer presented in Fig. 5. Thereafter, we focused on TDOS with addition of one adatom (shown in Fig. 5) and with those for 25 Li/Na adatoms on SnS\(_2\) included as supplementary material.
The pristine SnS\(_2\) monolayer in Fig. 5 is an insulator with a band gap of 2.44 eV measured between the valence band maximum (VBM) and conduction band minimum (CBM), indicating its deficiency for electronic conductivity. The TDOS of one Li/Na adatom on the H\(_{SnS_2}\) site of pristine SnS\(_2\) shown in Fig. 5c, e show that the Fermi level shifts towards the CBM, in agreement with the previous studies [64, 66, 67]. This reveals a possibility of electronic charges transferred (doping) into the system, and bringing up required electronic conductivity feature. Figure 5 presents the TDOS of SnS\(_2\) monolayer with S-vacancy. It is noted that the effect of S-vacancy maintains the insulating feature with a notable band gap, measuring at 2.37 eV. Just like for pristine SnS\(_2\) monolayer, an addition of Li/Na atom introduces electronic states within the band gap arising from CBM to the VBM (see Fig. 5d, f). As an additional result, note that the pristine and S-vacancy SnS\(_2\) monolayers display no magnetic properties, with the spin-up and spin-down density of states being identical. The addition of Li/Na adatoms does not impose the spin polarisation on the material.
Looking at 25 Li/Na adatoms on H\(_{SnS_2}\) site of both pristine and SnS\(_2\) monolayer with S-vacancy, the Fermi level shifts further into the CBM and more occupied states become populated within the band gap as shown in (Supplementary Fig. 2). This suggests that a modified SnS\(_2\) monolayer with topological defect can work well for LIBs.
Specific capacity and open circuit voltage

The calculated open circuit voltage versus specific capacity for the pristine and S-vacancy SnS\(_2\) monolayer with respective adatoms a Li and b Na increasing. The maximum specific capacity is obtained at the lowest voltage
Another important property to evaluate the performance of the SnS\(_2\) monolayer as an anode material is the open circuit voltage (V\(_{OC}\)). The V\(_{OC}\) represents the maximum voltage available from a cell, when no active current is flowing. It is derived by looking at the intercalation of adatoms onto the anode material, which decreases as intercalation increases. Thus, to obtain V\(_{OC}\) for Li/Na adatoms on the SnS\(_2\) monolayer, first consider the chemical reaction process for binding Li/Na adatoms on the substrate. These reactions are described by;
with x being the number of atoms undergoing intercalation during the reaction. Following this chemical reaction, we would have a change in V\(_{OC}\), which will in turn cause a change in the total energy of the cell. For a metallic adatom anode, where the chemical potential of the adatoms is constant, the electrical energy produced by intercalation is equal to the Gibbs free energy \(G_r\). The relationship between V\(_{OC}\) and \(G_r\) is;
where n is the number of adatoms in total adsorbed onto the SnS\(_2\) monolayer, and Faraday’s constant F is present as a conversion factor between joule (J), the standard units of energy for Gibbs free energy, and voltage V. The change in Gibbs free energy \(G_r\) is described by the equation;
where \(\Delta {E_r}\) is the change in internal energy, P is pressure, \(\Delta {V_r}\) is the change in volume, T is temperature, and \(\Delta {S_r}\) is the change in entropy. We approximate the calculations by assuming they occur at 0 K to further simplify the equation. As a result, we have that the term \(P\Delta {V_r}\) will be in the order of \(10^{-5}\) eV, much smaller than the order of magnitude for \(E_r\). Similarly, the \(T\Delta {S_r}\) term is of an order with thermal energy, which is also negligible at 0 K. We are left with \(\Delta {E_r}\), which has the same magnitude as the binding energy such that \(\Delta {E_r}=-\Delta {E_b}\). With the approximation we can use the following result [71];
We have that Faraday’s constant is absent, as the existing measurements for E\(_b\) are already in units of eV, making the inclusion of F for calculating V\(_{OC}\) unnecessary. Some conversion from eV to V must still occur through the cancellation of the electron charge e, though for eV we have that e is 1. The formula we have now obtained is near identical to Eq. (1) binding energy E\(_{b}\), bearing mostly a difference of units;
Unlike Eq. (1), it must be noted that \(E_{ad}\) refers to the bulk Li/Na energy calculated from the bulk body centred cube (BCC) lattice. Looking at Eq. (5), it is clear that V\(_{OC}\) will decrease simultaneously with binding energy as the number of adatoms increases, supported by Fig. 6a, b depicting our voltage as function of storage capacity for Li and Na adatoms respectively. It is generally known that for the maximum number of adatoms required on the SnS\(_2\) monolayer, the V\(_{OC}\) should approach 0.00 V. Just like for binding energy analysis, the positive V\(_{OC}\) values reveal that Li/Na adatoms on the SnS\(_2\) monolayer are likely to bound, while the negative values reveal undesired clustering. It is shown on Fig. 6a, b that the V\(_{OC}\) value for Li (Na) reduces from 2.51 V (2.95 V) to 1.34 V (1.68 V) as the storage capacity increases to the measured maximum on the 5\(\times\)5\(\times\)1 supercell for the pristine SnS\(_2\) monolayer. It is also noted that the Li/Na adatoms on SnS\(_2\) monolayer with S-vacancy have constantly high V\(_{OC}\) compared to pristine, for the entire plot. Qualitatively, the V\(_{OC}\) for Li (Na) reduces from 3.41 eV (3.80 eV) to 1.41 eV (1.70 eV) as the capacity increases to the measured maximum. The open circuit voltage average for Li (Na) on a pristine SnS\(_2\) monolayer is 2.06 V (2.03 V) and that on SnS\(_2\) monolayer with S-vacancy is 2.16 V (2.24 V). These values are within the acceptable range of 0.25–3.00 V [35, 51, 52]. As a result, these material systems will be able to yield significant charging voltage, and these V\(_{OC}\) values also indicate that the formation of dendrites can surely be averted during the battery operation.
The theoretical storage capacity C can be calculated using Eq. (3) below [71];
where z is the number of valence electrons of the adatoms, F is the Faraday constant with the value of 26,801 mAh/mol, and \(M_{SnS_2}\) is the total molar mass of the 5\(\times\)5\(\times\)1 supercell for the pristine SnS\(_2\) monolayer. The SnS\(_2\) monolayer with S-vacancy would have a slightly lower molar mass, and consequently, a slightly higher storage capacity if we consider Eq. (5). It is shown in Fig. 6a, b that the storage capacity value at the maximum number of Li adatoms on a pristine SnS\(_2\) monolayer (SnS\(_2\) monolayer with S-vacancy) is 1172.65 mAh/g (1195.95 mAh/g) and that of Na adatoms is 879.49 mAh/g (896.96 mAh/g). SnS\(_2\) has been determined to have a theoretical capacity of 1200 mAh/g for Li and 600 mAh/g for Na [35, 40, 72]. Practically, the maximum storage capacity is determined at a point where the voltage reaches 0.00 V, since by that point there will be no potential for the binding of additional adatoms [40, 66, 69, 73].
Conclusion
Using DFT approach, we systematically performed a comparative study of the adsorption and diffusion behaviour of Li/Na on a pristine SnS\(_2\) monolayer and on a SnS\(_2\) monolayer with S-vacancy for enhancement of electrochemical performance. We looked for improvements in the Li/Na binding energies, V\(_{OC}\), storage capacities and electronic conductivity. Although all adsorption sites are exothermic, it was established that Li/Na adatoms prefer to bind strongly on SnS\(_2\) monolayer with S-vacancy but avoiding the S-vacancy site (there is possible unwanted repulsive force between Li/Na and its surrounding Sn atoms ions). It was also noted that a great number of Li/Na ions can be stored on the substrate without forming bulk metallic, due to the steepness of binding energy plot. The Na ions with large atomic radii than Li ions have low binding energies. We also studied the diffusion of Li/Na across the SnS\(_2\) monolayer with S-vacancy by calculating activation energies, and it was established that avoiding the S-vacancy site along the path, an excellent diffusion barriers of 0.19 eV for Li and 0.13 eV for Na were achieved.
The effect of S-vacancy introduces some electronic states leading to slight shrinkage of the band gap from 2.44 eV to approximately 2.37 eV. An addition of Li/Na atoms further introduces electronic states on the band gap from the CBM to the VBM, and the induced states become more populated as the number of Li/Na adatoms increases, suggesting the possibility of enhanced electronic transmission performance which is a requirement for LIBs or SIBs. In this study, the calculated V\(_{OC}\) values of our electrodes materials are higher than those of graphite and graphene electrodes, owing to S atoms having more electronegativity than C atoms. It is also noted that the Li/Na adatoms on SnS\(_2\) monolayer with S-vacancy (created reactive sites) have high V\(_{OC}\) values compared to its pristine counterparts. The obtained V\(_{OC}\) values also indicate that the formation of dendrites can surely be averted for the envisaged battery operation. Understanding the effects of an S-vacancy on the electrochemical properties of Li/Na on the SnS\(_2\) monolayer allows us to consider possible improvements to energy storage devices that can be applied as a result of our improved anode material. Thus, opening the door to experimental studies into the viability of S-vacancies or vacancies in general in similar 2D materials.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Tarascon J, Armand M (2001) Issues and challenges facing rechargeable lithium batteries. Nature 414(6861):359–367
Vikström H, Davidsson S, Höök M (2013) Lithium availability and future production outlooks. Appl Energy 110:252–266
Roberts S, Kendrick E (2018) The re-emergence of sodium ion batteries: testing, processing, and manufacturability. Nanotechnol Sci Appl 11:23–33
Wang C, Sheng L, Jiang M, Lin X, Wang Q, Guo M, Wang G, Zhou X, Zhang X, Shi J, Jiang L (2023) Flexible SnSe[CDATA[_2]]/N-doped porous carbon-fiber film as anode for high-energy-density and stable sodium-ion batteries. J Power Sources 555:232405
Guo B, Wang X, Fulvio PF, Chi M, Mahurin SM, Sun XG, Dai S (2011) Soft-templated mesoporous carbon-carbon nanotube composites for high performance lithium-ion batteries. Adv Mater 23(40):4661–4666
Winter M, Besenhard JO, Spahr ME, Novák P (1998) Insertion electrode materials for rechargeable lithium batteries. Adv Mater 10(10):725–763
Andrew RC, Mapasha RE, Ukpong AM, Chetty N (2012) Mechanical properties of graphene and boronitrene. Phys Rev B 85
Chen KS, Balla I, Luu NS, Hersam MC (2017) Emerging opportunities for two-dimensional materials in lithium-ion batteries. ACS Energy Lett 2(9):2026–2034
Li H, Shi Y, Chiu MH, Li LJ (2015) Emerging energy applications of two-dimensional layered transition metal dichalcogenides. Nano Energy 18:293–305
Mortazavi B, Javvaji B, Shojaei F, Rabczuk T, Shapeev AV, Zhuang X (2021) Exceptional piezoelectricity, high thermal conductivity and stiffness and promising photocatalysis in two-dimensional MoSi[CDATA[_2]]N[CDATA[_4]] family confirmed by first-principles. Nano Energy 82:105716
Peng L, Zhu Y, Chen D, Ruoff RS, Yu G (2016) Two-dimensional materials for beyond-lithium-ion batteries. Adv Energy Mater 6(11):1600025
Novoselov KS, Geim AK, Morozov SV, Jiang D, Zhang Y, Dubonos SV, Grigorieva IV, Firsov AA (2004) Electric field effect in atomically thin carbon films. Science 306(5696):666–669
Al Hassan M, Sen A, Zaman T, Mostari M (2019) Emergence of graphene as a promising anode material for rechargeable batteries: a review. Mater Today Chem 11:225–243
Li L, Zhang D, Deng J, Gou Y, Fang J, Cui H, Zhao Y, Cao M (2021) Carbon-based materials for fast charging lithium-ion batteries. Carbon 183:721–734
Fan X, Zheng W, Kuo JL (2012) Adsorption and diffusion of li on pristine and defective graphene. ACS Appl Mater Interfaces 4(5):2432–2438
Fan X, Zheng WT, Kuo JL, Singh DJ (2013) Adsorption of single li and the formation of small li clusters on graphene for the anode of lithium-ion batteries. ACS Appl Mater Interfaces 5(16):7793–7797
Lee E, Persson KA (2012) Li absorption and intercalation in single layer graphene and few layer graphene by first principles. Nano Lett 12(9):4624–4628
Liu Y, Artyukhov VI, Liu M, Harutyunyan AR, Yakobson BI (2013) Feasibility of lithium storage on graphene and its derivatives. J Phys Chem Lett 4(10):1737–1742
Song J, Ouyang B, Medhekar NV (2013) Energetics and kinetics of li intercalation in irradiated graphene scaffolds. ACS Appl Mater Interfaces 5(24):12968–12974
Datta D, Li J, Koratkar N, Shenoy VB (2014) Enhanced lithiation in defective graphene. Carbon 80:305–310
Wu ZS, Ren W, Xu L, Li F, Cheng HM (2011) Doped graphene sheets as anode materials with superhigh rate and large capacity for lithium ion batteries. ACS Nano 5(7):5463–5471
Chua CK, Sofer Z, Khezri B, Webster RD, Pumera M (2016) Ball-milled sulfur-doped graphene materials contain metallic impurities originating from ball-milling apparatus: their influence on the catalytic properties. Phys Chem Chem Phys 18:17875–17880
Kawai S, Saito S, Osumi S, Yamaguchi S, Foster AS, Spijker P, Meyer E (2015) Atomically controlled substitutional boron-doping of graphene nanoribbons. Nat Commun 6
Ullah S, Denis PA, Sato F (2017) Beryllium doped graphene as an efficient anode material for lithium-ion batteries with significantly huge capacity: A dft study. Appl Mater Today 9:333–340
Wang H, Maiyalagan T, Wang X (2012) Review on recent progress in nitrogen-doped graphene: Synthesis, characterization, and its potential applications. ACS Catal 2(5):781–794
Cai K, Jing X, Zhang Y, Li L, Lang X (2022) A novel reed-leaves like aluminum-doped manganese oxide presetting sodium-ion constructed by coprecipitation method for high electrochemical performance sodium-ion battery. Int J Energy Res 46(10):14570–14580
Hartman T, Sofer Z (2019) Beyond graphene: Chemistry of group 14 graphene analogues: Silicene, germanene, and stanene. ACS Nano 13(8):8566–8576
Ivanovskii AL (2012) Graphene-based and graphene-like materials. Russ Chem Rev 81(7):571–605
Naguib M, Mashtalir O, Carle J, Presser V, Lu J, Hultman L, Gogotsi Y, Barsoum MW (2012) Two-dimensional transition metal carbides. ACS Nano 6(2):1322–1331
Shi M, Zhu H, Chen C, Jiang J, Zhao L, Yan C (2023) Synergistically coupling of graphene quantum dots with Zn-intercalated MnO[CDATA[_2]] cathode for high-performance aqueous Zn-ion batteries. Int J Min Metall Mater 30(1):25–32
Wang H, Chen Y, Yu H, Liu W, Kuang G, Mei L, Wu Z, Wei W, Ji X, Qu B, Chen L (2022) A Multifunctional Artificial Interphase with Fluorine-Doped Amorphous Carbon layer for Ultra-Stable Zn Anode. Adv Func Mater 32(43):2205600
Wang M, Jiang C, Zhang S, Song X, Tang Y, Cheng HM (2018) Reversible calcium alloying enables a practical room-temperature rechargeable calcium-ion battery with a high discharge voltage. Nat Chem 10(6):667–672
Zhang X, Tang Y, Zhang F, Lee CS (2016) A Novel Aluminum-Graphite Dual-Ion Battery. Adv Energy Mater 6(11):1502588
Bacaksiz C, Cahangirov S, Rubio A, Senger RT, Peeters FM, Sahin H (2016) Bilayer sns\(_2\): Tunable stacking sequence by charging and loading pressure. Phys Rev B 93
Samad A, Noor-A-Alam M, Shin YH (2016) First principles study of a sns2/graphene heterostructure: a promising anode material for rechargeable na ion batteries. J Mater Chem A 4:14316–14323
Dubrovskii GB (1998) Crystal structure and electronic spectrum of sns2. Phys Solid State 40(9):1557–1562
Xie R, Hu M, Liu D, Yu Y, Li C, He J, Luo J (2020) Atomic observation of phase transition in layered sns2 driven by in situ heating and electron beam irradiation. Appl Phys Lett 117(16):163103
Chang K, Wang Z, Huang G, Li H, Chen W, Lee JY (2012) Few-layer sns2/graphene hybrid with exceptional electrochemical performance as lithium-ion battery anode. J Power Sources 201:259–266
Zhu J, Yang D, Yin Z, Yan Q, Zhang H (2014) Graphene and graphene-based materials for energy storage applications. Small 10(17):3480–3498
Wang Y, Zhou J, Wu J, Chen F, Li P, Han N, Huang W, Liu Y, Ye H, Zhao F, Li Y (2017) Engineering sns2 nanosheet assemblies for enhanced electrochemical lithium and sodium ion storage. J Mater Chem A 5:25618–25624
Wei Z, Wang L, Zhuo M, Ni W, Wang H, Ma J (2018) Layered tin sulfide and selenide anode materials for li- and na-ion batteries. J Mater Chem A 6:12185–12214
Dubrovskii GB, Shelykh AI (1998) Photoelectric properties of tin disulfide. Phys Solid State 40(8):1295–1296
Huang Y, Sutter E, Sadowski JT, Cotlet M, Monti OL, Racke DA, Neupane MR, Wickramaratne D, Lake RK, Parkinson BA, Sutter P (2014) Tin disulfide-an emerging layered metal dichalcogenide semiconductor: materials properties and device characteristics. ACS Nano 8(10):10743–10755
Guan D, Li J, Gao X, Yuan C (2015) Carbon nanotube-assisted growth of single-/multi-layer sns2 and sno2 nanoflakes for high-performance lithium storage. RSC Adv 5. https://doi.org/10.1039/C5RA09613H
Chong P, Zhou Z, Li Y, Wang J, Xiu J, Wei M (2023) Three-dimensional structure S-SnS\(_2\)/NSG with sulfur vacancies for high-performance lithium-ion batteries. J Alloys Compd 939
Su J, Pei Y, Yang Z, Wang X (2014) Ab initio study of graphene-like monolayer molybdenum disulfide as a promising anode material for rechargeable sodium ion batteries. RSC Adv 4:43183–43188
Jiang Y, Song D, Wu J, Wang Z, Huang S, Xu Y, Chen Z, Zhao B, Zhang J (2019) Sandwich-like sns2/graphene/sns2 with expanded interlayer distance as high-rate lithium/sodium-ion battery anode materials. ACS Nano 13(8):9100–9111
Liu Z, Deng H, Mukherjee PP (2015) Evaluating pristine and modified sns2 as a lithium-ion battery anode: A first-principles study. ACS Appl Mater Interfaces 7(7):4000–4009
Ma Z, Yao Z, Cheng Y, Zhang X, Guo B, Lyu Y, Wang P, Li Q, Wang H, Nie A, Aspuru-Guzik A (2020) All roads lead to rome: Sodiation of different-stacked sns2. Nano Energy 67:104276
Zhang J, Cao D, Wu Y, Cheng X, Kang W, Xu J (2020) Phase transformation and sulfur vacancy modulation of 2d layered tin sulfide nanoplates as highly durable anodes for pseudocapacitive lithium storage. Chem Eng J 392:123722
Huang Y, Ding S, Xu S, Ma ZF, Yuan X (2022) Binder-free sns2 sheet array with high sulfur vacancy concentration for enhanced lithium storage performance. Electrochim Acta 409:139979
Wang J, Huang J, Huang S, Notohara H, Urita K, Moriguchi I, Wei M (2020) Rational design of hierarchical sns2 microspheres with s vacancy for enhanced sodium storage performance. ACS Sustain Chem Eng 8(25):9519–9525
Capelle K (2002) A bird’s-eye view of density-functional theory. Braz J Phys 36. https://doi.org/10.1590/S0103-97332006000700035
Blöchl P, Först C, Schimpl J (2003) Projector augmented wave method: Ab initio molecular dynamics with full wave functions. Bull Mater Sci 26:33–41. https://doi.org/10.1007/BF02712785
Giannozzi P, Baroni S, Bonini N, Calandra M, Car R, Cavazzoni C, Ceresoli D, Chiarotti GL, Cococcioni M, Dabo I, Corso AD, de Gironcoli S, Fabris S, Fratesi G, Gebauer R, Gerstmann U, Gougoussis C, Kokalj A, Lazzeri M, Martin-Samos L, Marzari N, Mauri F, Mazzarello R, Paolini S, Pasquarello A, Paulatto L, Sbraccia C, Scandolo S, Sclauzero G, Seitsonen AP, Smogunov A, Umari P, Wentzcovitch RM (2009) QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J Phys: Condens Matter 21(39)
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865–3868
Monkhorst HJ, Pack JD (1976) Special points for brillouin-zone integrations. Phys Rev B 13:5188–5192
Fu CL, Ho KM (1983) First-principles calculation of the equilibrium ground-state properties of transition metals: applications to nb and mo. Phys Rev B 28:5480–5486
Blöchl PE, Jepsen O, Andersen OK (1994) Improved tetrahedron method for brillouin-zone integrations. Phys Rev B 49:16223–16233
Grimme S, Antony J, Ehrlich S, Krieg H (2010) A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 132(15):154104
Jónsson H, Mills G, Jacobsen KW (1998) Classical and Quantum Dynamics in Condensed Phase Simulations, World Scientific, chap Nudged elastic band method for finding minimum energy paths of transitions, pp. 385–404.https://www.worldscientific.com/doi/abs/10.1142/9789812839664_0016
Lv Y, Chen B, Zhao N, Shi C, He C, Li J, Liu E (2016) Interfacial effect on the electrochemical properties of the layered graphene/metal sulfide composites as anode materials for li-ion batteries. Surf Sci 651:10–15
Liu Y, Hu B, Kong D, Wang J, Li Y, Su J, Xia C (2019) Effect of S-vacancy on the oxidation state of Ce in monolayer SnS[CDATA[_2]] Int J Mod Phys B 33(26):1950308. https://doi.org/10.1142/S0217979219503089
Mukherjee S, Banwait A, Grixti S, Koratkar N, Singh CV (2018) Adsorption and diffusion of lithium and sodium on defective rhenium disulfide: A first principles study. ACS Appl Mater Interfaces 10(6):5373–5384
Wu H, Guo Z, Zhou J, Sun Z (2019) Vacancy-mediated lithium adsorption and diffusion on mxene. Appl Surf Sci 488:578–585
Makaremi M, Mortazavi B, Rabczuk T, Ozin GA, Singh CV (2019) Theoretical investigation: 2d n-graphdiyne nanosheets as promising anode materials for li/na rechargeable storage devices. ACS Appl Nano Mater 2(1):127–135
Zhou Y, Geng C (2017) A MoO[CDATA[_2]] sheet as a promising electrode material: ultrafast Li-diffusion and astonishing Li-storage capacity. Nanotechnology 28(10):105402
Wang F, Yao Q, Zhou L, Ma Z, He M, Wu F (2018) Theoretical understanding of sns monolayer as li ion battery anode material. J Phys Chem Solids 121:261–265
Huang T, Tian B, Guo J, Shu H, Wang Y, Dai J (2019) Semiconducting borophene as a promising anode material for li-ion and na-ion batteries. Mater Sci Semicond Process 89:250–255
Rao YC, Yu S, Gu X, Duan XM (2019) Prediction of MoO2 as high capacity electrode material for (Na, K, Ca)-ion batteries. Appl Surf Sci 479:64–69
Aydinol MK, Kohan AF, Ceder G, Cho K, Joannopoulos J (1997) Ab initio study of lithium intercalation in metal oxides and metal dichalcogenides. Phys Rev B 56:1354–1365
Kong Z, Huang M, Liang Z, Tu H, Zhang K, Shao Y, Wu Y, Hao X (2022) Phosphorus doping induced the co-construction of sulfur vacancies and heterojunctions in tin disulfide as a durable anode for lithium/sodium-ion batteries. Inorg Chem Front 9:902–913
Rehman J, Fan X, Zheng W (2019) Computational insight of monolayer sns2 as anode material for potassium ion batteries. Appl Surf Sci 496
Acknowledgements
The authors are grateful to the University of Pretoria for financial support and computational resources. REM is very appreciative to National Institute for Theoretical and Computational Sciences (NITheCS) platform for financial support. The authors are also grateful to Centre of High Performance Computing (CHPC) initiatives based in Cape Town for supplying them with large hardware computing facilities to expediently carry out the calculations.
Funding
Open access funding provided by University of Pretoria.
Author information
Authors and Affiliations
Contributions
C.A. Bekeur conceptualised this work, and performed all the calculations, analyses, writing and proofreading. R.E. Mapasha conceptualised this work, and performed analyses of results, writing and proofreading.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bekeur, C.A., Mapasha, R. Enhancement of electrochemical performance of monolayer SnS\(_2\) for Li/Na-ion batteries through a sulphur vacancy: a DFT study. J Solid State Electrochem 27, 2445–2456 (2023). https://doi.org/10.1007/s10008-023-05518-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-023-05518-6