
Abstract
Nafion offers microporous channels of typically 1–4-nm diameter for cation exchange. Recently, it has been shown that these cation exchanger properties can be inverted to allow anion binding by pre-filling the hydrophilic channel structure. Here, pre-filling is performed with hydrous iron oxide and sensitivity towards anionic phosphate and arsenate analytes is investigated. After a period of phosphate/arsenate accumulation, the voltammetric response in aqueous 1 M NaNO3 is obtained based on the Fe(III/II) redox process. The position of the peak at distinct potentials clearly reveals the presence of either phosphate or arsenate, presumably present in the form of FePO4 and FeAsO4. In the presence of mixtures of phosphate and arsenate, a competition of FePO4 versus FeAsO4 nucleation (within the Nafion microporous host) is suggested to result in a switch of phosphate detection at higher concentrations (ca. > 500 μM, solubility controlled) towards arsenate detection at lower concentrations (ca. < 500 μM, nucleation controlled). This phenomenon is suggested to be linked to the Ostwald step rule.

Hydrous iron oxide embedded into Nafion is shown to give distinct voltammetric responses after reaction with either phosphate or arsenate with a switch from growth control to nucleation control at low concentration
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Detection and removal of arsenite/arsenate are important in ground and well water treatment [1] and have often been achieved with absorbents such as those based on hydrous iron oxide materials [2], magnetite nano-materials [3], or nano-composites such as graphene-ferric oxide [4, 5], and ferrates [6]. Also, chitosan-based bio-sorbents have been developed [7] and polymer-inorganic hybrids [8, 9]. A review on arsenate binding onto nano-adsorbents has appeared [10]. Examples for analytical detection of As(V) or As(III) have been developed based on adsorbents like ferric oxyhydroxide [11] with x-ray photoelectron spectroscopy (XPS) or based on hydrous ferric hydroxide with x-ray fluorescence (XRF) [12]. A highly sensitive method has been reported based on enhanced Raman on thiol-modified silver nanoparticles [13, 14]. The removal of arsenate with the help of hydrous iron oxide [15] or with mixed oxides [16,17,18] has been reported and exploited. There is a strong link from the thermodynamics of solid-phase formation to the sorption ability for ions to solid surfaces, e.g. on minerals or on ferric oxide surfaces [19]. Therefore, the solubility product is often considered to be the key parameter in predicting arsenate or phosphate removal (or detection). In certain cases (as shown in this report for phosphate and arsenate), effects introduced by the rate of nucleation may also be important and this can then lead to the binding of the more soluble component even in the presence of the less soluble component.
Voltammetric detection of arsenite and arsenate has often been based on adsorption onto oxide surfaces. The adsorption of arsenite on rutheninum nano-oxides was employed [20] and a method based on Fe3O4 was reported [21]. A liquid|liquid arsenate transfer voltammetry method has been investigated [22] as a way to distinguish/separate arsenate and phosphate. A stripping voltammetry method based on nano-MnFe2O4 has been developed [23]. The role of Mn as catalyst in As(V) conversion/detection has been highlighted by work by van den Berg [24]. As(III) and/or As(V) detection have been reported also with noble metal catalysts, for example, based on voltammetry at gold-modified boron-doped diamond [25], with platinum nanoparticles [26, 27], at gold nanoparticles [28], at electro-aggregated gold in phosphate media [29], at gold-palladium nanoparticles [30], and at other types of bimetallic nanoparticles [31].
The reaction of arsenate with ferric cations is used to remove the toxic metal from water streams [32] and groundwater [33], usually aided by electrocoagulation processes. However, both arsenate and phosphate react in a similar way. Competitive adsorption of arsenate onto haematite has been studied [34] and a very strong inhibition of arsenate binding in the presence of phosphate was noted (consistent with the thermodynamically preferred formation of the phosphate product). It is interesting to re-visit this competitive binding of phosphate and arsenate in the context of voltammetric analysis at modified electrodes. In the current study, competitive adsorption with hydrous iron oxide immobilised into the micropore channels in commercial Nafion is investigated at screen-printed carbon electrodes.
Nafion is a technical ionomer material with a micro-channel structure of typically 1–4-nm diameter [35,36,37] (Fig. 1). Binding of highly negatively charged anions such as phosphate and arsenate into a semi-permeable ionomer material such as Nafion [38] would usually be ruled out. However, recently, it has been shown that the negatively charged channels of Nafion can be pre-filled for example with cationic iron complexes to result in a binding and sensor function even for anions including arsenite [39]. The cationic species immobilised in the negatively charged ion channel can balance out charges and effectively open the Nafion ion channels to anions. This observation led to the initial idea for the use of Fe(III) within Nafion to react with solution-phase anions such as phosphate or arsenate. Figure 1 shows a schematic drawing to illustrate the strategy followed in this report: initial filling of Nafion with Fe(III), which is then converted to hydrous iron oxide, to then allow anion binding and detection for phosphate and for arsenate.

Schematic drawing of the use of Nafion ionomer to (i) accumulate aqueous Fe3+ into the nano-channel strucutre, then (ii) to expose to 0.1 M NaOH to form hydrous iron oxide in the microporous channels. When exposed to phosphate or arsenate (here indicated as A−), accumulation of the anion into the Nafion micropore channel structure occurs
The hydrous iron oxide immobilised within the channels inverts the cation-exchanging properties of Nafion to allow anion binding within or in the vicinty of the host. Hence, sensitivity to anionic analytes can be investigated, as arsenate or phosphate ions can be taken up within these channels. A voltammetric response is obtained indirectly (for the Fe(III/II) redox system) to indicate distinct peaks for phosphate or for arsenate. It is shown that competition in binding of phosphate or arsenate occurs (case A) at higher concentrations to give the more insoluble iron phosphate, but (case B) it occurs at lower concentration to give the more readily nucleating iron arsenate.
Experimental
Chemical reagents
Nafion (5 wt% in alcohol), iron(III) nitrate, nitric acid, sodium hydroxide, sodium phosphate (di-basic), sodium arsenate (di-basic), and sodium nitrate were obtained from Sigma-Aldrich and used without further purification. Solutions were prepared (0.01 M Fe(III) nitrate, 1 M NaNO3, 0.1 M sodium hydroxide, 10 mM stock solution of sodium arsenate and sodium phosphate) in deionised and filtered water with 18.2 Ωcm resistivity (at 22 °C).
Instrumentation
A microAutoLab II Potentionstat (Metrohm) was employed for cyclic voltammetry experiments employing a conventional three-electrode configuration. Screen-printed electrodes were employed (DropSense DS 110) with a central carbon disk, a carbon counter, and a speudo-silver reference electrode.
Procedures
Electrode preparation
A Nafion coating was applied by dipping of screen-printed electrodes into commercial Nafion solution followed by air-drying. Next, the modified electrode was dipped into 10 mM Fe(III) nitrate solution (pH 3) and left immersed for 5 min. The electrode is then rinsed with water and air-dried to give a yellow colouration. Next, the electrode is dipped into 0.1 M NaOH and left immersed for 5 min. The colouration changes to orange. Rinsing with water and air-drying at 80 °C give the electrode ready to store or use in experiments.
Analysis protocol
The modified electrode was dipped into arsenate/phosphate solutions for 25-min accumulation without potential control. Next, the electrode is rinsed and transferred to an electrochemical cell with 1.0 M NaNO3 electrolyte. Voltammetric experiments are performed either in cyclic voltammetry or in linear sweep voltammetry mode by scanning the applied potential from a positive potential into the Fe(III/II) reduction region.
Results and discussion
Formation and characterisation of hydrous Fe(III) oxide–Nafion film electrodes
Initial studies were performed with Nafion film deposits on polished glassy carbon substrates, but the surface morphology was observed to be too smooth for a thin Nafion deposit to stay intact and for a multi-step experimental procedure to be feasible. Film deposits were particularly unstable when exposed to aqueous Fe3+. Therefore, screen-printed carbon electrodes with higher surface roughness were employed for the Nafion film deposition and further experimental steps.
The screen-printed carbon electrode was fully immersed into 5 wt% Nafion (in ethanol) and then removed and dried to give an evenly coated film (see the “Experimental” section). Figure 2a, b shows the scanning electron microscopy (SEM) image of the electrode before and after Nafion coating (and with further treatments, see below). The elements fluorine and sulphur are indicative for the presence of the Nafion ionomer coating. Next, the modified electrode was dipped into aqueous 10 mM Fe(III) nitrate (at pH 3) for 5 min to allow cation exchange and accumulation of Fe(III) within the Nafion ion exchanger (visible by eye as a yellow colouration). This electrode is then rinsed with water and air-dried followed by dipping into aqueous 0.1 M NaOH for 5 min. A colour change from yellow to orange indicates formation of hydrous iron oxide probably within the microporous channels in Nafion. After a rinse with water and drying at 80 °C, the electrode is ready for use. Figure 2b shows SEM images for this type of electrode before exposure to arsenate solution and Fig. 2c shows the electrode after exposure to arsenate (0.1 mM for 25 min). The elemental analysis (from energy dispersive x-ray analysis) shows As at a level about 25% by weight relative to that of Fe. The presence of the polymer is linked to a haze in the image and limits definition for the underlying carbon features.

Scanning electron microscopy (SEM) images for a bare screen-printed carbon electrode (a), the electrode coated with Nafion with embedded hydrous iron oxide (b), and the electrode after immersion into arsenate solution (c) (see text)
Further characterisation was performed for flakes of material from the electrode surface with transmission electron microscopy (TEM) to provide better details at the nanometre scale (Fig. 3). Figure 3a, b shows a Nafion fragment of approximately 1-μm diameter. When focusing on even smaller details, only faint structural features are resolved (Fig. 3c), but the elemental composition suggests the presence of Nafion. In Fig. 3d, e, the effect of Fe(III) absorption and conversion to hydrous iron oxide is demonstrated. Small regions of darker colouration appear indicative of the inorganic components. Finally, after reaction with arsenate (Fig. 3f), smaller dark grains are visible and the elemental signature for As is observed.

Transmission electron microscopy (TEM) images for a, b a Nafion fragment; c Nafion with elemental composition; d, e hydrous iron oxide in Nafion; and f arsenate-treated hydrous iron oxide in Nafion with elemental composition
Electrochemical response to phosphate or arsenate with hydrous Fe(III) oxide–Nafion film electrodes
The screen-printed carbon electrode modified with Nafion and hydrous iron oxide was then exposed to a test solution containing 1 mM arsenate in water to investigate the voltammetric response. Instead of pursuing the in situ voltammetry, a more reproducible method was developed in which the electrode is first exposed to a test solution without control of the electrode potential. Next, the electrode is transferred into aqueous NaNO3 solution for voltammetry. Electrochemical processes within ionomer environments are sensitive to the ionic strength of the supporting electrolyte solution and therefore initially a series of test experiments were conducted to explore this parameter.
The voltammetric response for the reduction of Fe(III) arsenate is observed at − 0.22 V versus SCE (see Fig. 4(iii)) but this cathodic peak is only reliably observed in the presence of 1 M NaNO3 (higher concentrations of NaNO3 did not improve the voltammetric response). In the presence of 0.1 M NaNO3 the magnitude of the peak halves and with 0.01 M NaNO3, it is not observed. Therefore, all following experiments are conducted in the presence of 1 M NaNO3 supporting electrolyte.

Cyclic voltammograms (scan rate 50 mV s−1) in 1.0 M NaNO3 for a screen-printed carbon electrode (i) with Nafion coating, (ii) with Nafion and hydrous iron oxide, and (iii) coated with Nafion and hydrous iron oxide after reaction with 1 mM arsenate for 25 min
Figure 4(i) shows the cyclic voltammogram for a screen-printed carbon electrode with only Nafion film deposit. With hydrous iron oxide embedded, two significant voltammetric responses are observed (Fig. 4(ii)). The chemically reversible Fe3+/Fe2+ signal with a midpoint potential of approximately 0.08 V versus SCE is indicative of some mobile iron species with the Nafion deposit (Eq. 1). A second reduction signal at − 0.43 V versus SCE could be assigned to another Fe(III) species possibly due to the hydrous oxide. After exposure of the screen-printed electrode to the arsenate test solution (1 mM), a new reduction peak is observed at − 0.22 V versus SCE and assigned here to the presence of an embedded Fe(III) arsenate (Eq. 2).
In the voltammetric response, any remaining Fe3+(Nafion) seems depleted. The formation of FeAsO4(Nafion) is likely to proceed from the outside of the Nafion (with arsenate diffusion to the interface) towards the inner nano-channels and therefore the electrochemical response may also be dominated by material formed close to the Nafion|electrolyte interface in the vicinity of the electrode. A similar experiment performed with 1 mM HPO42− in the test solution does result in a different peak response (see Fig. 5(ii)) at a potential of − 0.48 V versus SCE clearly distinguished from the FeAsO4 signal. A corresponding process (Eq. 3) is suggested to explain the voltammetric signal.

Cyclic voltammograms (scan rate 50 mV s−1) for a screen-printed carbon electrode immersed in 1 M NaNO3 for (i) FeAsO4 embedded in Nafion and (ii) FePO4 embedded in Nafion
The more negative potential for the voltammetric reduction is likely to be linked to the energy required for the lattice energy of the solid to be overcome. Therefore, a more negative reduction peak would be consistent with lower solubility. The reported solubility products for FePO4 (9.92 × 10−29) and for FeAsO4 (10−23 [40]) are consistent with this interpretation. However, other hydrated crystal forms for both solid iron phosphate and iron arsenate are known, and therefore interpretation of data based on these solubility products has to be qualitative. However, the clear separation of phosphate and arsenate peak responses suggests that analysis based on voltammetry over a given concentration range should be possible. Next, the competition in the formation of either FePO4 or FeAsO4 is further investigated.
Electrochemical response to phosphate and arsenate with hydrous Fe(III) oxide–Nafion film electrodes
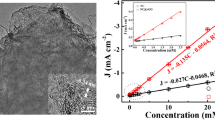
When performing voltammetric measurements for HAsO42− as a function of concentration, initially, the time for the screen-printed electrode immersed in the test solution needed to be investigated. In particular for lower arsenate concentrations, shorter exposure did not result in sufficient conversion for voltammetric signals to be obtained. Long exposure of up to 60 min was employed to obtain voltammetric signals even at a 1 μM HAsO42− level. As a compromise between experimentation time and sensitivity here, an exposure time of 25 min was selected. Figure 6 shows typical voltammetric peaks for the Fe(III/II) process recorded with linear sweep voltammetry (scan rate 50 mV s−1). Data are baseline corrected (with a second-order polynomial) and inverted to give a positive peak signal. The peak current is then plotted versus concentration of arsenate. A linear relationship is observed for the range from 10 to 1000 μM, but perhaps intriguingly an initial peak produces an offset from zero current.

Linear sweep voltammograms (scan rate 50 mV s−1; processed with second-order polynomial baseline subtraction and shown as positive peak) in 1 M NaNO3 for arsenate detection with a screen-printed carbon electrode coated with Nafion and hydrous iron oxide. Plot (A) shows the peak current versus arsenate concentration and plot (B) shows the peak current versus phosphate concentration
A similar set of experiments with phosphate test solutions demonstrates a similar behaviour over a similar range of concentrations, but with a peak response at a more negative potential. The linear plots in Fig. 6(A, B) summarise the effect of concentration on the signal indicating that concentration and signal are positively correlated.
Next, competition experiments with both phosphate and arsenate present in equal concentration (in the same solution) were performed. Data are presented in Fig. 7. At higher concentrations (keeping PO43− and AsO43− concentrations equal), the phosphate signal is observed, implying that phosphate anions are taken up by the Nafion microporous channels more easily. When increasing the concentration, larger amounts of Fe(III) phosphate product appear to form and hence an increase in the voltammetric signal is observed. However, at lower concentrations, more specifically at concentrations lower than 400–500 μM, a switch occurs and arsenate ions are taken up by the immobilised hydrous iron oxide more readily than phosphate. Hence, a switch from phosphate peaks to arsenate peaks is evident with no intermediate case where both phosphate and arsenate peaks are seen together. Figure 7(B) clearly shows the switch in the peak potential. This observation suggests that at lower concentrations, FeAsO4 is formed preferentially to FePO4 with significant implications for arsenate removal and detection.

Data from linear sweep voltammograms (scan rate 50 mV s−1; obtained in aqueous 1 M NaNO3; processed with second-order polynomial baseline subtraction and shown as positive peak) for simultaneous phosphate arsenate detection with a screen-printed carbon electrode coated with Nafion and hydrous iron oxide. Plot (A) shows the peak current versus arsenate/phosphate concentration in water and plot (B) shows the peak potential versus arsenate/phosphate concentration in water
The processes at higher concentration appear to be consistent with the solubility product for the phosphate being dominant. Therefore, FePO4 is the thermodynamically preferred product. However, at a lower concentration, kinetic control can prevail and the formation of FeAsO4 appears to be the faster process and therefore the dominating reaction. Cases of competitive nucleation kinetics are well-known [41] and important, for example in the Ostwald step rule [42]. Intermediate “kinetic” products (with higher solubility) nucleate faster and form in processes in which ultimately the thermodynamic product (with lower solubility) would be formed. For the case of hydrous iron oxide immobilised into the micropores of Nafion and under conditions employed in this study, the preferential/faster nucleation of FeAsO4 appears to suppress the formation of FePO4 and therefore results in the voltammetric response for arsenic at lower concentrations (see Fig. 8). These observations are preliminary in nature and more work will be required to confirm this effect for a wider range of conditions. However, control over selectivity in the removal and detection of arsenate will be of interest in particular when based on materials such as hydrous iron oxide.

Schematic drawing indicating the effect of phosphate/arsenate concentration on the competitive nucleation of FePO4 and FeAsO4 at higher and lower concentrations, respectively
Conclusions
Nafion-coated screen-printed electrodes can be used to indirectly detect both phosphate and arsenate anions accumulated from electrolyte solutions. The electrodes are disposable and need first to be immersed into the test solution. Then, after transfer into the 1 M NaNO3 measurement solution, voltammetric signals are observed. For both phosphate and arsenate, a Fe(III/II) reduction process is responsible for the voltammetric peak. There is a linear relationship between the concentration of the anion and the magnitude of the signal. The formation of FePO4 or FeAsO4 within the vicinity of Nafion micropore channels is proposed. Competitive nucleation of these two materials is proposed to cause a switch from phosphate detection to arsenate binding when going from higher to lower concentration. Further work will be required to better unravel the parameters that contribute to this selectivity for arsenate (e.g. presence of electrolyte, pH, microporous host material, reaction time).
For future analytical applications, the sensitivity of the method would have to be significantly improved (namely reaching below 10 μg dm−3 As corresponding to 0.13 μM in agreement with WHO recommendations [43]). Also, the switch in detection of phosphate versus arsenate needs to be better understood to allow measurements for drinking water quality to be quantified for both arsenate and phosphate. Finally, other types of redox active absorbents for competitive binding of phosphate and arsenate could be developed for application in Nafion or in other similar microporous host materials.
References
Cullen WR, Reimer KJ (1989) Arsenic speciation in the environment. Chem Rev 89(4):713–764
Bissen M, Frimmel FH (2003) Arsenic—a review. Part II: oxidation of arsenic and its removal in water treatment. Acta Hydrochim Hydrobiol 31(2):97–107
Liu CH, Chuang YH, Chen TY, Tian Y, Li H, Wang MK, Zhang W (2015) Mechanism of arsenic adsorption on magnetite nanoparticles from water: thermodynamic and spectroscopic studies. Environm Sci Technol 49(13):7726–7734
Andjelkovic I, Tran DNH, Kabiri S, Azari S, Markovic M, Losic D (2015) Graphene aerogels decorated with alpha-FeOOH nanoparticles for efficient adsorption of arsenic from contaminated waters. ACS Appl Mater Interfaces 7(18):9758–9766
Zhu J, Lou ZM, Liu Y, Fu RQ, Baig SA, Xu XH (2015) Adsorption behavior and removal mechanism of arsenic on graphene modified by iron-manganese binary oxide (FeMnOx/RGO) from aqueous solutions. RSC Adv 5(83):67951–67961
Sharma VK, Zboril R, Varma RS (2015) Ferrates: greener oxidants with multimodal action in water treatment technologies. Acc Chem Res 48(2):182–191
Boddu VM, Abburi K, Talbott JL, Smith ED, Haasch R (2008) Removal of arsenic(III) and arsenic(V) from aqueous medium using chitosan-coated biosorbent. Water Res 42(3):633–642
DeMarco MJ, Sengupta AK, Greenleaf JE (2003) Arsenic removal using a polymeric/inorganic hybrid sorbent. Water Res 37(1):164–176
Padungthon S, German M, Wiriyathamcharoen S, Sengupta AK (2015) Polymeric anion exchanger supported hydrated Zr(IV) oxide nanoparticles: a reusable hybrid sorbent for selective trace arsenic removal. React Funct Polym 93:84–94
Ray PZ, Shipley HJ (2015) Inorganic nano-adsorbents for the removal of heavy metals and arsenic: a review. RSC Adv 5(38):29885–29907
Ding M, de Jong BHWS, Roosendaal SJ, Vredenberg A (2000) XPS studies on the electronic structure of bonding between solid and solutes: adsorption of arsenate, chromate, phosphate, Pb2+, and Zn2+ ions on amorphous black ferric oxyhydroxide. Geochim Cosmochim Acta 64(7):1209–1219
Hettipathirana TD, Smith LH, Norrish K (2001) Simultaneous determination of low parts-per-billion level Pb and As in waters using energy-dispersive x-ray fluorescence spectrometry. Appl Spectrosc 55:7028–7032
Li JL, Chen LX, Lou TT, Wang YQ (2011) Highly sensitive SERS detection of As3+ ions in aqueous media using glutathione functionalized silver nanoparticles. ACS Appl Mater Interfaces 3(10):3936–3941
Mulvihill M, Tao A, Benjauthrit K, Arnold J, Yang P (2008) Surface-enhanced Raman spectroscopy for trace arsenic detection in contaminated water. Angew Chem Int Ed 47(34):6456–6460
Wu K, Liu RP, Li T, Liu HJ, Peng JM, Qu JH (2013) Removal of arsenic(III) from aqueous solution using a low-cost by-product in Fe-removal plants-Fe-based backwashing sludge. Chem Eng J 226:393–401
Zhang GS, Qu JH, Liu HJ, Liu RP, Wu RC (2007) Preparation and evaluation of a novel Fe-Mn binary oxide adsorbent for effective arsenite removal. Water Res 41(9):1921–1928
Zhang SX, Niu HY, Cai YQ, Zhao XL, Shi YL (2010) Arsenite and arsenate adsorption on coprecipitated bimetal oxide magnetic nanomaterials: MnFe2O4 and CoFe2O4. Chem Eng J 158(3):599–607
Zhang GS, Liu FD, Liu HJ, Qu JH, Liu RP (2014) Respective role of Fe and Mn oxide contents for arsenic sorption in iron and manganese binary oxide: an X-ray absorption spectroscopy investigation. Environm Sci Technol 48(17):10316–10322
Sadiq M (1997) Arsenic chemistry in soils: an overview of thermodynamic predictions and field observations. Water Air Soil Pollut 93(1–4):117–136
Gupta R, Gamare JS, Pandey AK, Tyagi D, Kamat JV (2016) Highly sensitive detection of arsenite based on its affinity toward ruthenium nanoparticles decorated on glassy carbon electrode. Anal Chem 88(4):2459–2465
Gao C, Yu XY, Xiong SQ, Liu JH, Huang XJ (2013) Electrochemical detection of arsenic(III) completely free from noble metal: Fe3O4 microspheres-room temperature ionic liquid composite showing better performance than gold. Anal Chem 85(5):2673–2680
Marken F, Hayman CM, Page PCB (2002) Phosphate and arsenate electro-insertion processes into a N,N,N′,N′-tetraoctylphenylenediamine redox liquid. Electrochem Commun 4(5):462–467
Zhou SF, Han XJ, Fan HL, Zhang QX, Liu YQ (2015) Electrochemical detection of As(III) through mesoporous MnFe2O4 nanocrystal clusters by square wave stripping voltammetry. Electrochim Acta 174:1160–1166
Gibbon-Walsh K, Salaün P, van den Berg CMG (2012) Determination of arsenate in natural pH seawater using a manganese-coated gold microwire electrode. Anal Chim Acta 710:50–57
Song Y, Swain GM (2007) Development of a method for total inorganic arsenic analysis using anodic stripping voltammetry and a Au-coated, diamond thin-film electrode. Anal Chem 79(6):2412–2420
Hrapovic S, Liu Y, Luong JHT (2007) Reusable platinum nanoparticle modified boron doped diamond microelectrodes for oxidative determination of arsenite. Anal Chem 79(2):500–507
Dai X, Compton RG (2006) Detection of As(III) via oxidation to As(V) using platinum nanoparticle modified glassy carbon electrodes: arsenic detection without interference from copper. Analyst 131(4):516–521
Dai X, Nekrassova O, Hyde ME, Compton RG (2004) Anodic stripping voltammetry of arsenic(III) using gold nanoparticle-modified electrodes. Anal Chem 76(19):5924–5929
Rassaei L, Sillanpää M, French RW, Compton RG, Marken F (2008) Arsenite determination in phosphate media at electroaggregated gold nanoparticle deposits. Electroanalysis 20(12):1286–1292
Lan YC, Luo HJ, Ren XH, Wang YP, Liu YZ (2012) Anodic stripping voltammetric determination of arsenic(III) using a glassy carbon electrode modified with gold-palladium bimetallic nanoparticles. Microchim Acta 178(1–2):153–161
Moghimi N, Mohapatra M, Leung KT (2015) Bimetallic nanoparticles for arsenic detection. Anal Chem 87(11):5546–5552
Delaire C, Amrose S, Zhang MH, Hake J, Gadgil A (2017) How do operating conditions affect As(III) removal by iron electrocoagulation? Water Res 112:185–194
van Genuchten CM, Addy SEA, Pena J, Gadgil AJ (2012) Removing arsenic from synthetic groundwater with iron electrocoagulation: an Fe and As K-edge EXAFS study. Environ Sci Technol 46(2):986–994
Yang GC, Liu YY, Song SX (2015) Competitive adsorption of As(V) with co-existing ions on porous hematite in aqueous solutions. J Environ Chem Engineer 3(3):1497–1503
Leddy J (2015) Modification of nafion on membranes: tailoring properties for function. In: Liu JL, Bashir S (eds) Nanomaterials for sustainable energy. ACS Symp Ser. 1213, pp 99–133
Mauritz KA, Moore RB (2004) State of understanding of nafion. Chem Rev 104(10):4535–4585
Davis TA, Pletcher D, Genders D (1997) A first course in ion permeable membranes. The Electrochemical Consultancy, Southampton
He DP, Madrid E, Aaronson BDB, Fan L, Doughty J, Mathwig K, Bond AM, McKeown NB, Marken F (2017) A cationic diode based on asymmetric Nafion film deposits. ACS Appl Mater Interfaces 9(12):11272–11278
Azad UP, Yadav DK, Ganesan V, Marken F (2016) Hydrophobicity effects in iron polypyridyl complex electrocatalysis within Nafion thin-film electrodes. Phys Chem Chem Phys 18(33):23365–23373
Langmuir D, Mahoney J, Rowson J (2006) Solubility products of amorphous ferric arsenate and crystalline scorodite (FeAsO4 · 2H2O) and their application to arsenic behavior in buried mine tailings. Geochim Cosmochim Acta 70(12):2942–2956
VanSsanten RA (1984) The Ostwald step rule. J Phys Chem 88(24):5768–5769
Ostwald W (1897) Studien über die bildung und umwandlung fester körper. Phys Chem 22:289–330
WHO Arsenic Fact Sheet accessed 30th December 2017 via http://www.who.int/mediacentre/factsheets/fs372/en/
Acknowledgements
N.G.N.A. and F.M. thank the Commonwealth Scholarship Commission for financial support (CMCF-2016-5) for collaboration between University of Dschang (Cameroon) and the University of Bath (UK).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sivasothy, T., Ndifor-Angwafor, N.G. & Marken, F. Voltammetric characteristics of hydrous Fe(III) oxide embedded into Nafion and immobilised onto a screen-printed carbon electrode: binding of arsenate versus phosphate. J Solid State Electrochem 22, 3059–3067 (2018). https://doi.org/10.1007/s10008-018-4009-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-018-4009-9