
Abstract
Hydrogen electrosorption was performed in thin electrodeposits of Pd alloys with Pt, Au, and Rh. The possibility of their application as phase charging–discharging systems was investigated. The values of specific pseudocapacitance, power, and energy were calculated for hydrogen-saturated Pd-rich electrodes for temperatures 283–313 K. The best working parameters are exhibited by Pd–Rh alloys with 85–95% Pd, and by Pd–Pt alloys with 90–95% Pd in the bulk. The maximum values of specific pseudocapacitance are ca. 4,500 F g−1, specific energy ca. 150 J g−1 and specific power up to 750 W g−1 (per the mass of the electroactive material). In the case of the alloy deposits on reticulated vitreous carbon, their characteristics related to the total mass of the electroactive material and the substrate are comparable with those for other supercapacitors utilizing various redox reactions.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Pd and its alloys represent a large class of hydrogen-absorbing materials [1], which are important for chemical/electrochemical power sources. These systems can be applied in batteries, fuel cells or for hydrogen storage [2, 3]. Moreover, since hydrogen can be electrochemically inserted into and extracted from Pd-based electrodes, such materials can be treated as phase charging–discharging systems for electrochemical capacitors. This type of devices usually utilizes the capacitance of electrical double layer of various carbon materials or pseudocapacitance connected with reversible redox processes like insertion of atomic species into the crystal structure of bulk solid electrodes, e.g. conducting polymers or transition metal oxides (e.g., RuO2, IrO2, MnO2) [4–7]. Due to high currents generated during hydrogen uptake and removal as well as their good practical reversibility, thin Pd-based layers electrolytically loaded with hydrogen seem particularly promising for that purpose.
Recently, we have reported on the possibility of the use of hydrogen-saturated Pd and Pd-rich Pd–Rh alloys (with 92–97% Pd in the bulk) deposited on reticulated vitreous carbon (RVC) in supercapacitors [8–10]. It was found that these systems exhibited good working parameters, with specific pseudocapacitance reaching 540 F g−1, specific energy up to 20 Wh kg−1 and specific power up to 6 W g−1. All these values were within the range reported for other types of supercapacitors [4–7].
In this work, we summarize the characteristics of Pd alloys with Pt, Au, and Rh saturated with absorbed hydrogen in the aspect of their possible application in electrochemical capacitors. We show the influence of temperature and alloy bulk composition on the values of specific pseudocapacitance, power, and energy. We demonstrate that supercapacitors based on Pd-rich Pd–Rh and Pd–Pt alloys are characterized by better working parameters than pure Pd. For the deposits on RVC substrate their characteristic are comparable with those for other supercapacitors utilizing various redox reactions.
Experimental
All experiments were performed in 0.5 M H2SO4 solutions at temperature of 283, 298 and 313 K, controlled by a thermostat (Lauda RE 306, Germany). The solutions were prepared from analytical grade reagents and triply distilled water additionally purified in a Millipore system. Before the experiments, the electrolyte was deoxygenated with an argon (99.999%) stream for 20 min; during the experiments, the argon stream was directed above the solution level in order to avoid contact with air. A Hg|Hg2SO4|0.5 M H2SO4 was used as the reference electrode. A Pt gauze was used as the auxiliary electrode. All potentials are recalculated with respect to the reversible hydrogen electrode (RHE) in the working solution.
The working electrode was a gold wire (99.99%, 0.5 mm diameter) covered with a thin alloy layer electrodeposited at a constant potential from a bath containing aqueous solutions of PdCl2 and HAuCl4, H2PtCl4 or RhCl3 with the addition of HCl. Various alloy compositions were obtained by changing bath composition and deposition potential. The alloy thickness was ca. 0.9 ± 0.2 μm. The details of the preparation of thin Pd-based deposits on Au substrate are given in other reports [8–10]. For comparison, the characteristic of Pd and Pd–Rh deposits (thickness in the range 0.6–1.0 μm) on RVC with the porosity grade of 20 pores per inch (ppi) is presented on the basis of our previous studies (see [11–13] for more information).
All alloy compositions are bulk compositions expressed in atomic percentages. After electrochemical measurements the electrodes were dissolved in aqua regia and the total amounts of the metals in the alloys were analyzed by atomic absorption spectroscopy. Additionally, alloy bulk compositions were determined utilizing the linear dependence of the potential of the phase transition between the α- and β-phase of absorbed hydrogen on Pd bulk content, as described in detail in our earlier communication [14].
At the beginning of hydrogen absorption experiments each electrode was subjected to a series of voltammetric and chronoamperometric scans/steps through the potential region of hydrogen adsorption and absorption until a steady state voltammogram was obtained. This procedure was applied in order to avoid the effects of aging during further hydrogen insertion/removal [15–17].
Hydrogen absorption was performed at potential −0.01 V vs. RHE for a period sufficient to ensure full hydrogen saturation, determined by chronoamperometry. At this potential, it was possible to load the electrodes with hydrogen in a short time with avoiding a significant hydrogen evolution. Hydrogen was desorbed under voltammetric (a single scan to 0.49 V vs. RHE at 0.01 V s−1) or chronoamperometric (a potential step to 0.49 V vs. RHE) conditions. Hydrogen extraction potential was adjusted to make possible a rapid removal of all absorbed hydrogen without the parallel oxidation of the electrode surface.
Results and discussion
Figure 1 shows typical voltammetric and chronoamperometric responses recorded during hydrogen desorption after earlier electrode saturation with hydrogen. These curves are typical of hydrogen extraction form thin Pd-based layers and were described in detail in earlier reports [12–17]. By the integration of hydrogen oxidation currents the amount of absorbed hydrogen was obtained. It was found that under the applied conditions of hydrogen sorption/desorption the oxidation of hydrogen from solution has no significant influence on the amount of absorbed hydrogen calculated from the hydrogen oxidation charge. It was calculated that the cathodic charge during hydrogen loading is not higher than ca. 5% in comparison with the anodic charge during the oxidation of absorbed hydrogen.

The amount of hydrogen absorbed in Pd-based materials can be recalculated into units commonly used for description of energy storage systems, i.e., pseudocapacitance per mass unit. Under voltammetric conditions, specific pseudocapacitance is obtained according to the equation:
where Q oxHabs is charge due to the oxidation of absorbed hydrogen, ΔE is the potential range in which absorbed hydrogen is completely oxidized, and m M is mass of the hydrogen-absorbing material (i.e., the mass of Pd or Pd alloy deposit). The parameters used in the calculation are illustrated in Fig. 1a.
The value of specific pseudocapacitance of the electrode saturated with hydrogen can be estimated in the following way. Absorbed hydrogen oxidation charge is given by the expression:
where n H is the amount of absorbed hydrogen, n M is the amount of the hydrogen-absorbing metal, M M is molar mass of the metal, d M is metal density, l M is the thickness of the metallic layer and S is the geometric area of a substrate for the metallic layer.
Pseudocapacitance per the unit of mass of the metal is equal to:
Taking into account the values of a maximum H/M ratio typical of a Pd layer of a thickness of 1 μm (i.e. H/M = 0.73) and ΔE typical of the voltammetric hydrogen extraction with a scan rate sufficient for the removal of all absorbed hydrogen in a single anodic scan (i.e. ΔE = 0.2 V, v = 0.01 V s−1) one obtains C = 3,300 F g−1 (per the mass of Pd deposit). Similar values were found experimentally for Pd layers with a thickness in the range of 0.6–1.0 μm [11–13].
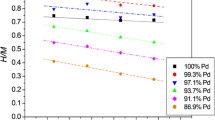
The calculations of specific pseudocapacitance according to Eq. 1 have been performed for hydrogen-saturated Pd–Pt, Pd–Au and Pd–Rh alloys. The changes in pseudocapacitance per deposit mass with alloy bulk compositions, at various temperatures, are shown in Fig. 2. It is visible that for Pd-rich Pd–Rh alloys the specific pseudocapacitance is higher than that for pure Pd, reaching a maximum for 88–95% Pd. For these alloy compositions, the pseudocapacitance is by ca. 40–50% greater than for pure Pd. This tendency mirrors the fact that Pd-rich Pd–Rh alloys are characterized by a greater hydrogen absorption capacity in comparison with Pd [10, 15]. Moreover, the width of hydrogen oxidation peak in voltammetry is reduced upon Pd alloying with Rh. The latter effect means that hydrogen can be extracted from Pd–Rh alloys in a narrower potential range that from pure Pd. Therefore, in the formula used for the calculation of specific pseudocapacitance (Eq. 3) the numerator not only increases, but also the denominator decreases, which leads to a greater increase in specific pseudocapacitance than the value expected on the basis of the changes in absorption capacity alone. The effect of peak width reduction is also observed for Pd–Pt alloys; however, in that case the amount of absorbed hydrogen is always lower than in pure Pd [16]. As a result, the increase in pseudocapacitance for Pd-rich Pd–Pt alloys is smaller than for Pd–Rh alloys and after a weaker maximum (pseudocapacitance by up to 25% greater than for Pd) for ca. 90–95% Pd, the capacitance markedly drops for lower Pd contents. In the case of Pd–Au alloys, the unfavorable effect of the decrease in absorption capacity dominates over the beneficial effect of Au addition on hydrogen peak width [17], which leads to a monotonic capacity loss with increasing Au bulk content.

Influence of temperature and alloy composition on specific pseudocapacitance (per mass of electroactive material) for hydrogen desorption process
In the temperature range studied, temperature has little influence on pseudocapacitance values. This is due to the fact that with increasing temperature both the amount of absorbed hydrogen (i.e. Q oxHabs in Eqs. 1–3) and the hydrogen peak width (i.e. ΔE) slightly decrease, resulting only in small changes in pseudocapacitance.
For the characteristic of a real charging/discharging system working as a supercapacitor the mass of a carrier has to be taken into account. In earlier papers [12, 13], we reported on the electrochemical behavior of Pd and Pd–Rh (92–97% Pd in the bulk) layers deposited on RVC. This material has been widely described in the literature as a promising electrode material with possible applications in electrochemical power sources [18–23]. In our experiments, the metal/alloy layers of a thickness of 0.6–1.0 μm were deposited on RVC with 20 ppi porosity grade. In that case, the total mass of the electrode material (m) is a sum of the hydrogen-absorbing layer (m M) and RVC substrate (m RVC):
where d RVC is RVC density (0.048 g cm−3), and S/V is the surface-to-volume ratio characteristic of RVC material of a given porosity grade (14 cm2 cm−3 for 20 ppi) [20]. Thus, the specific pseudocapacitance for H–Pd/RVC system can be estimated from the equation:
Taking into account that for Pd deposits on RVC the width of hydrogen oxidation peak is greater (ΔE′ = 0.5 V) than for Pd deposited on Au wire, while the H/M ratio is practically unchanged, one obtains the values of C = 340 F g−1 for 1 μm layer of Pd on RVC (20 ppi). The experimental values found in our studies [11–13] were in the range 240–430 F g−1 for Pd and up to 540 F g−1 for Pd–Rh alloys (97% Pd in the bulk). This tendency is consistent with the data presented in Fig. 2. It is noteworthy that the values of specific pseudocapacitance for hydrogen-saturated Pd-based systems are comparable with the data reported for supercapacitors based on reversible insertion of atomic species into bulk solid electrodes, e.g., polymers or metal oxides, and higher than the values typical of double layer electrochemical capacitors [4–7].
Other parameters used for the characterization of supercapacitors are specific power and specific energy. Specific energy can be obtained as:
An average value of specific power can be obtained under conditions of chronoamperometric hydrogen oxidation according to the relation:
where Q oxHabs is charge due to the oxidation of absorbed hydrogen, t is a period required for a complete hydrogen desorption and Ε abs − Ε des is the difference between absorption and desorption potentials in chronoamperometric experiments (here 0.50 V).
For desorption time typically of the order of 1 s and H/M = 0.73 one obtains the value of specific energy and average specific power for pure Pd ca. 130 J g−1 and 330 W g−1, respectively. By the analogy to specific pseudocapacitance (Eq. 5) taking into account the influence of the RVC substrate the values of specific energy and power can be expressed as follows:
where ΔE′ is the potential range in which absorbed hydrogen is completely oxidized from a Pd deposit on RVC (here ca. 0.5 V), while t is a period required for a complete hydrogen desorption from Pd deposit on RVC (ca. 25 s for Ε abs − Ε des = 0.50 V). Equations 5, 8, and 9 allow the estimation of the values of the working parameters for these systems deposited on RVC (note, that in Eq. 8 E denotes specific energy, but ΔE stands for potential difference).
Additionally, maximum specific power accessible during hydrogen extraction can be calculated from the maximum value of hydrogen oxidation current (i max), i.e. at the very beginning of hydrogen extraction in a chronoamperometric experiment (see Fig. 1b):
The influence of alloy bulk composition on the values of specific power and energy (per mass of the deposit) for Pd alloys with Au, Pt or Rh is presented in Figs. 3 and 4. It is demonstrated that for Pd-rich Pd–Pt alloys both average and maximum values of specific power exceed those for pure Pd, with a maximum corresponding to Pd bulk content around 90%. For these alloy compositions, the specific power at 313 K is almost by 100% higher than for pure Pd. This behavior is a consequence of the positive effect of Pt addition to Pd on the kinetics of hydrogen electrosorption, which is mirrored in higher current and shorter time corresponding to hydrogen oxidation [16]. For other alloys, average specific power is not higher than for Pd. However, for Pd–Rh alloys the values of maximum specific power are intermediate between those for Pd and Pd–Pt alloys, with a maximum between 90% and 95% Pd. It should be noted again that for the Pd–Rh system the tendencies are in line with those observed earlier for the deposits on RVC, where the average values of specific power were ca. 3 W g−1 for Pd/RVC and ca. 2 W g−1 for Pd–Rh/RVC [13], while maximum specific power reached 6 W g−1. For all systems, specific power increases with temperature due to the acceleration of hydrogen electrosorption process [16, 17].

Influence of temperature and alloy composition on specific energy (per mass of electroactive material) for hydrogen desorption process

Influence of temperature and alloy composition on: a average specific power, b maximum specific power (per mass of electroactive material) for hydrogen desorption process
The values of specific energy decrease with increasing content of the alloying metal and with increasing temperature. In the case of Pd and Pd–Rh deposits on RVC, the values of specific energy reached ca. 80 J g−1 [13].
Figure 5 shows the changes in the values of specific pseudocapacitance as well as average and maximum specific power during repeated hydrogen absorption/desorption under cyclic voltammetric conditions at 298 K for Pd and a Pd–Rh alloy (92.6% Pd in the bulk) deposited on Au wires. This procedure was applied to check charging–discharging durability of a potential supercapacitor. No significant deterioration of hydrogen absorption capacity was observed even after 800 cycles. The changes in the specific pseudocapacitance between maximum and minimum values did not exceed 5%. It means that during prolonged cyclic loading–deloading procedure no irreversible oxidation of the electrode material or metal losses occurred. The values of specific power even raised slightly with the progress of cycling due to the improvement in the kinetics of hydrogen electrosorption. The latter effect was reported in the literature [24] and was explained by a successive creation of defects in Pd crystal lattice which facilitate further transport of hydrogen in the metal.

Variation of specific pseudocapacitance and power with the number of hydrogen absorption/desorption cycles (scan rate 0.01 V s−1) in the potential region between −0.01 and 0.49 V (298 K) for Pd and a Pd–Rh alloy (92.6% Pd in the bulk)
Data presented in Figs. 2, 3 and 4 lead to the conclusion that hydrogen-saturated Pd-rich (in the range of 85–95% Pd in the bulk) Pd–Pt and Pd–Rh alloys are promising electrode materials to be utilized in supercapacitors. Both these systems are characterized by higher pseudocapacitance and maximum specific power with respect to the Pd–H system. Additionally, Pd–Pt alloys exhibit also higher average specific power as compared to Pd.
Conclusions
Hydrogen-saturated Pd alloys with Pt, Au and Rh can be applied as phase charging–discharging systems for electrochemical capacitors. The best specific pseudocapacitance is exhibited by Pd–Rh alloys with 85–95% Pd, and by Pd–Pt alloys with 90–95% Pd in the bulk. These systems are also characterized by higher specific power than Pd. When these alloys are deposited on RVC substrate, their characteristics are comparable with those typical of supercapacitors utilizing various redox reactions.
References
Lewis FA (1967) The palladium–hydrogen system. Academic, London
Kleperis J, Wójcik G, Czerwiński A, Skowroński J, Kopczyk M, Bełtowska-Brzezinska M (2001) J Solid State Electrochem 5:229–249
Schlapbach L, Züttel A (2001) Nature 414:353–358
Zhang Y, Feng H, Wu X, Wang L, Zhang A, Xia T, Dong H, Li X, Zhang L (2009) Int J Hydrog Energy 34:4889–4899
Obreja VVN (2008) Phys E 40:2596–2605
Huggins RA (2000) Solid State Ionics 134:179–195
Lokhande CD, Dubal DP, Joo OS (2011) Curr Appl Phys 11:255–270
Łukaszewski M, Czerwiński A (2010) Thin Solid Films 518:3680–3689
Łukaszewski M, Hubkowska K, Czerwiński A (2011) J Electroanal Chem 651:131–142
Koss U, Łukaszewski M, Hubkowska K, Czerwiński A (2011) J Solid State Electrochem 15:2477–2487
Czerwiński A, Łukaszewski M, Żurowski A, Patent RP 204948
Czerwiński A, Łukaszewski M, Żurowski A, Siwek H, Obrębowski S (2006) J New Mat Elect Syst 9:419–429
Łukaszewski M, Żurowski A, Czerwiński A (2008) J Power Sources 185:1598–1604
Łukaszewski M, Żurowski A, Grdeń M, Czerwiński A (2007) Electrochem Commun 9:671–676
Żurowski A, Łukaszewski M, Czerwiński A (2006) Electrochim Acta 51:3112–3117
Hubkowska K, Łukaszewski M, Czerwiński A (2011) Electrochim Acta 56:2344–2350
Hubkowska K, Łukaszewski M, Czerwiński A (2010) Electrochim Acta 56:235–242
Czerwiński A, Dmochowska M, Grdeń M, Kopczyk M, Wójcik G, Młynarek G, Kołata J, Skowroński JM (1999) J Power Sources 77:28–33
Wang J (1981) Electrochim Acta 26:1721–1726
Friedrich JM, Ponce-de-León C, Reade GW, Walsh FC (2004) J Electroanal Chem 561:203–217
Rogulski Z, Lewdorowicz W, Tokarz W, Czerwiński A (2004) Pol J Chem 78:1357–1370
Czerwiński A, Rogulski Z, Obrębowski S, Siwek H, Paleska I, Chotkowski M, Łukaszewski M (2009) J Appl Electrochem 39:559–567
Czerwiński A (1995) Pol J Chem 69:699–706
Millet P, Srour M, Faure R, Durand R (2001) Electrochem Commun 3:478–482
Acknowledgments
This work was financially supported by NCN (National Center of Science), grant UMO-2011/01/B/ST4/00442 (Warsaw University: 120000-504/66-4003).
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Łukaszewski, M., Hubkowska, K., Koss, U. et al. Characteristic of hydrogen-saturated Pd-based alloys for the application in electrochemical capacitors. J Solid State Electrochem 16, 2533–2539 (2012). https://doi.org/10.1007/s10008-012-1658-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-012-1658-y