
Abstract
Context
Hydrogen bonds (HB) influence the conformational preferences of biomolecules and their optical and electronic properties. The directional interaction of molecules of water can be a prototype to understand the effects of HBs on biomolecules. Among the neurotransmitters (NT), L-aspartic acid (ASP) stands out due to its importance in health and as a precursor of several biomolecules. As it presents different functional groups and readily forms inter- and intramolecular HBs, ASP can be considered a prototype for understanding the behavior of NTs when interacting by HB with other substances. Although several theoretical studies have been performed in the past on isolated ASP and its formed complexes with water, both in gas and liquid phases, using DFT and TD-DFT formalisms, these works did not perform large basis set calculations or study electronic transitions of ASP-water complexes. We investigated the HB interactions in complexes of ASP and water molecules. The results show that the interactions between the carboxylic groups of ASP with water molecules, forming cyclic structures with two HBs, lead to more stable and less polar complexes than other conformers formed between water and the NH2 group. It was observed that there is a relationship between the deviation in the UV–Vis absorption band of the ASP and the interactions of water with the HOMO and LUMO orbitals with the stabilization/destabilization of the S1 state to the S0 of the complexes. However, in some cases, such as 1:1 complex ASP-W2, this analysis may be inaccurate due to small changes in ΔE.
Methods
We studied the landscapes of the ground state surface of different conformers of isolated L-ASP and the L-ASP-(H2O)n complexes (n = 1 and 2) using the DFT formalism, with the B3LYP functional, and six different basis sets: 6–31 + + G(d,p), 6–311 + + G(d,p), D95 + + (d,p), D95V + + (d,p), cc-pVDZ, and, cc-pVTZ basis sets. The cc-pVTZ basis set provides the minimum energy of all conformers, and therefore, we performed the analysis with this basis set. We evaluated the stabilization of the ASP and complexes using the minimum ground state energy, corrected by the zero point energy and the interaction energy between the ASP and the water molecules. We also calculated the vertical electronic transitions S1 ← S0, and their properties using the TD-DFT formalism at B3LYP/cc-pVTZ level with the optimized geometries for S0 state with the same basis set. For the analysis of the vertical transitions of isolated ASP and the ASP-(H2O)n complexes, we calculated the electrostatic energy in the S0 and S1 states. We performed the calculations with the Gaussian 09 software package. We used the VMD software package to visualize the geometries and shapes of the molecule and complexes.





Similar content being viewed by others

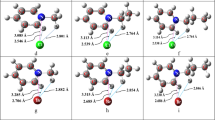
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Arunan E, Desiraju GR, Klein RA et al (2011) Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl Chem 83:1637–1641. https://doi.org/10.1351/PAC-REC-10-01-02
Schran C, Marsalek O, Markland T (2017) Unravelling the influence of quantum proton delocalization on electronic charge transfer through the hydrogen bond. Chem Phys Lett 678:289–295. https://doi.org/10.1016/j.cplett.2017.04.034
Deng W, You Y, Zhang A (2015) Supramolecular network-based self-healing polymer materials. In: Recent advances in smart self-healing polymers and composites. Woodhead Publishing, pp 181–210. https://doi.org/10.1016/B978-1-78242-280-8.00007-8
Buckingham AD, del Bene JE, McDowell SAC (2008) The hydrogen bond. Chem Phys Lett 463:1–10. https://doi.org/10.1016/j.cplett.2008.06.060
Herschlag D, Pinney MM (2018) Hydrogen bonds: simple after all? Biochemistry 57:3338–3352. https://doi.org/10.1021/acs.biochem.8b00217
Anju LS, Aruldhas D, Hubert Joe I, Balachandran S (2020) Density functional theory, spectroscopic and hydrogen bonding analysis of fenoxycarb–water complexes. J Mol Struct 1201:127201. https://doi.org/10.1016/j.molstruc.2019.127201
Sakti AW, Nishimura Y, Nakai H (2020) Recent advances in quantum-mechanical molecular dynamics simulations of proton transfer mechanism in various water-based environments. Wiley Interdiscip Rev Comput Mol Sci 10:e1419. https://doi.org/10.1002/wcms.1419
da Cruz VV, Gel’mukhanov F, Eckert S et al (2019) Probing hydrogen bond strength in liquid water by resonant inelastic X-ray scattering. Nat Commun 10:1–9. https://doi.org/10.1038/s41467-019-08979-4
Khatib R, Backus EHG, Bonn M et al (2016) Water orientation and hydrogen-bond structure at the fluorite/water interface. Sci Rep 6:1–10. https://doi.org/10.1038/srep24287
Nelson DL, Cox MM (2017) Lehninger principles of biochemistry, 7th edn. Freeman, New York, W.H
Guria S et al (2019) Rapid detection of aspartic acid and glutamic acid in water by BODIPY-based fluorescent probe: Live-cell imaging and DFT studies. Dyes Pigm 168:111–122. https://doi.org/10.1016/j.dyepig.2019.04.052
Topo E, Soricelli A, D’Aniello A et al (2009) The role and molecular mechanism of D-aspartic acid in the release and synthesis of LH and testosterone in humans and rats. Reprod Biol Endocrinol 7:120. https://doi.org/10.1186/1477-7827-7-120
Katane M, Homma H (2011) d-Aspartate—an important bioactive substance in mammals: a review from an analytical and biological point of view. J Chromatogr B 879:3108–3121. https://doi.org/10.1016/j.jchromb.2011.03.062
Amantea D, Bagetta G (2017) Excitatory and inhibitory amino acid neurotransmitters in stroke: from neurotoxicity to ischemic tolerance. Curr Opin Pharmacol 35:111–119. https://doi.org/10.1016/j.coph.2017.07.014
Chen M, Lin Z (2007) Ab initio studies of aspartic acid conformers in gas phase and in solution. J Chem Physics 127:154314. https://doi.org/10.1063/1.2777161
Nakayoshi T, Kato K, Fukuyoshi S et al (2020) Computational studies on nonenzymatic succinimide-formation mechanisms of the aspartic acid residues catalyzed by two water molecules. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics 1868(9):140459. https://doi.org/10.1016/j.bbapap.2020.140459
Wang H, Huang Z, Shen T, Guo L (2012) Hydrogen-bonding interactions in adrenaline-water complexes: DFT and QTAIM studies of structures, properties, and topologies. J Mol Model 18:3113–3123. https://doi.org/10.1007/s00894-011-1325-8
Oda A, Nakayoshi T, Fukuyoshi S et al (2018) Influences of conformations of peptides on stereoinversions and/or isomerizations of aspartic acid residues. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics 1866:783–788. https://doi.org/10.1016/j.bbapap.2018.01.006
Kato K, Nakayoshi T, Kurimoto E, Oda A (2019) Computational studies on the nonenzymatic deamidation mechanisms of glutamine residues. ACS Omega 4:3508–3513. https://doi.org/10.1021/acsomega.8b03199
Becke AD (2014) Perspective: Fifty years of density-functional theory in chemical physics. J Chem Phys 140:8A301. https://doi.org/10.1063/1.4869598
Zheng YZ, Deng G, Guo R et al (2019) A DFT-based study of the hydrogen-bonding interactions between myricetin and ethanol/water. J Mol Model 25:1–10. https://doi.org/10.1007/s00894-019-3940-8
Alam MJ, Ahmad S (2012) Anharmonic vibrational studies of l-aspartic acid using HF and DFT calculations. Spectrochim Acta A Mol Biomol Spectrosc 96:992–1004. https://doi.org/10.1016/j.saa.2012.07.135
Bhunia S, Srivastava SK, Materny A, Ojha AK (2016) A vibrational and conformational characterization of arginine at different pH values investigated using Raman spectroscopy combined with DFT calculations. J Raman Spectrosc 47:1073–1085. https://doi.org/10.1002/jrs.4918
Frish MJ, Pople JA, Binkley JS (1984) Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J Chem Phys 80:3265–3269. https://doi.org/10.1063/1.447079
He LJ, Fan JF, Tang M (2010) DFT study of gaseous conformers of aspartic acid. J Theor Comput Chem 9:687–700. https://doi.org/10.1142/S021963361000592X
Petersson GA, Al-Laham MA (1991) A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J Chem Phys 94:6081–6090. https://doi.org/10.1063/1.460447
Dunning Jr. TH, Hay PJ (1977) Modern theoretical chemistry. In: Schaffer HF III (ed) 2. Plenum Press, New York
Barbieri PL, Fantin PA, Jorge FE (2006) Gaussian basis sets of triple and quadruple zeta valence quality for correlated wave functions. Mol Phys 104:2945–2954. https://doi.org/10.1080/00268970600899018
Dunning TH (1989) Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J Chem Phys 90:1007–1023. https://doi.org/10.1063/1.456153
Papajak E, Leverentz HR, Zheng J, Truhlar DG (2009) Efficient diffuse basis sets: Cc-pVxZ+ and maug-cc-pVxZ. J Chem Theory Comput 5:1197–1202. https://doi.org/10.1021/ct800575z
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb, MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M,, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr. JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2016) Gaussian 09, Revision A.02. Gaussian Inc., Wallingford, CT
Humphrey W, Dalke A, Schulten K (1996) VMD: Visual molecular dynamics. J Mol Graph 14:33–38. https://doi.org/10.1016/0263-7855(96)00018-5
Tiwari A, Honingh C, Ensing B (2019) Accurate calculation of zero point energy from molecular dynamics simulations of liquids and their mixtures. J Chem Physics 151:244124. https://doi.org/10.1063/1.5131145
Csonka GI, Ruzsinszky A, Perdew JP (2005) Estimation, computation, and experimental correction of molecular zero-point vibrational energies. J Phys Chem A 109:6779–6789. https://doi.org/10.1021/jp0519464
Buemi G (2006) Intramolecular hydrogen bonds. methodologies and strategies for their strength evaluation. In: Grabowski S (ed) Hydrogen bonding - new insights. Springer, Netherlands, New York, pp 51–107
Jensen F (2017) Introduction to computational chemistry, 3rd ed. John Wiley & Sons, Ltd, San Francisco
Richard RM, Bakr BW, Sherrill CD (2018) Understanding the many-body basis set superposition error: beyond boys and Bernardi. J Chem Theory Comput 14:2386–2400. https://doi.org/10.1021/acs.jctc.7b01232
Boys SF, Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys 19:553–566. https://doi.org/10.1080/00268977000101561
Anikina EV, Beskachko VP (2020) Basis set superposition error: effects of atomic basis set optimization on value of counterpoise correction. Bulletin of the South Ural State University. Ser. Mathematics. Mechanics Physics 12(1):55–62. https://doi.org/10.14529/mmph200107
Gray M, Bowling PE, Herbert JM (2022) Systematic evaluation of counterpoise correction in density functional theory. J Chem Theory Comput. https://doi.org/10.1021/acs.jctc.2c00883
Lesiuk M, Jeziorski B (2019) Size consistency and counterpoise correction in explicitly correlated calculations of interaction energies and interaction-induced properties. Phys Rev A Coll Park 99:032712. https://doi.org/10.1103/PhysRevA.99.032712
Adamo C, Jacquemin D (2013) The calculations of excited-state properties with time-dependent density functional theory. Chem Soc Rev 42:845–856. https://doi.org/10.1039/c2cs35394f
Israelachvili JN (2011) Intermolecular and surface forces, 3rd ed. Academic Press, Massachusetts
Mulliken RS (1955) Electronic population analysis on LCAO–MO molecular wave functions. J Chem Phys 23:1833–1840. https://doi.org/10.1063/1.1740588
Demircioʇlu Z, Kaştaş ÇA, Büyükgüngör O (2015) Theoretical analysis (NBO, NPA, Mulliken Population Method) and molecular orbital studies (hardness, chemical potential, electrophilicity and Fukui function analysis) of (E)-2-((4-hydroxy-2-methylphenylimino)methyl)-3-methoxyphenol. J Mol Struct 1091:183–195. https://doi.org/10.1016/J.MOLSTRUC.2015.02.076
Miar M, Shiroudi A, Pourshamsian K et al (2021) Theoretical investigations on the HOMO–LUMO gap and global reactivity descriptor studies, natural bond orbital, and nucleus-independent chemical shifts analyses of 3-phenylbenzo[d]thiazole-2(3H)-imine and its para-substituted derivatives: Solvent and subs. J Chem Res 45:147–158. https://doi.org/10.1177/1747519820932091
Velardez GF, Ferrero JC, Beswick JA, Daudey JP (2001) Ab initio study of the structures and π* ← n electronic transition in formic acid−(water) n (n = 3, 4, and 5) hydrogen bonded complexes. J Phys Chem A 105:8769–8774. https://doi.org/10.1021/jp0100295
Aikens CM, Gordon MS (2006) Incremental solvation of nonionized and zwitterionic glycine. J Am Chem Soc 128:12835–12850. https://doi.org/10.1021/ja062842p
Kagra D, Preethi SP, Sharma P (2020) Interaction of aspartic acid and asparagine with RNA nucleobases: a quantum chemical view. J Biomol Struct Dyn 38:943–955. https://doi.org/10.1080/07391102.2019.1592025
Alonso JL, Cocinero EJ, Lesarri A et al (2006) The Glycine-Water Complex. Angew Chem 118:3551–3554. https://doi.org/10.1002/ange.200600342
Rai AK, Xu X, Lin Z, Rai DK (2011) Conformational search for zwitterionic leucine and hydrated conformers of both the canonical and zwitterionic leucine using the DFT-CPCM model. Vib Spectrosc 56:74–81. https://doi.org/10.1016/j.vibspec.2010.10.006
Altis A, Nguyen PH, Hegger R, Stock G (2007) Dihedral angle principal component analysis of molecular dynamics simulations. J Chem Physics 126:244111. https://doi.org/10.1063/1.2746330
Glusker JP (1998) Directional aspects of intermolecular interactions. Design of organic solids. Springer, Berlin, Heidelberg, pp 1–56
Shimoni L, Glusker JP (1995) Hydrogen bonding motifs of protein side chains: descriptions of binding of arginine and amide groups. Protein Sci 4:65–74. https://doi.org/10.1002/pro.5560040109
Pavia DL, Lampam GM, Kriz G, Vyviam JR (2014) Introduction to spectroscopy, 5th ed. Cengage Learning, Stanford
Bredas JL (2014) Mind the gap! Mater Horiz 1:17–19. https://doi.org/10.1039/C3MH00098B
Silva AM, Silva P, Sales FAM et al (2012) Optical absorption and DFT calculations in L-aspartic acid anhydrous crystals: charge carrier effective masses point to semiconducting behavior. Phys Rev B 86:19520. https://doi.org/10.1103/PhysRevB.86.195201
Gilli G, Gilli P (2009) The nature of the hydrogen bond: outline of a comprehensive hydrogen bond theory, 1st edn. Oxford University Press, New York
Zhang M, Zhang M, Zhao G (2018) Time-dependent density functional theory (TDDFT) study on the electronic spectroscopic blue-shift phenomenon and photoinduced charge transfer of firefly luciferin anion in aqueous solution: Insight into the excited-state hydrogen bond weakening mechanism. J Lumin 195:116–119. https://doi.org/10.1016/j.jlumin.2017.11.015
Zhao GJ, Han KL (2012) Hydrogen bonding in the electronic excited state. Acc Chem Res 45:404–413. https://doi.org/10.1021/ar200135h
Li X, Liu L, Schlegel H (2002) Bernhard. On the physical origin of blue-shifted hydrogen bonds. J Am Chem Soc 124:9639–9647. https://doi.org/10.1021/ja020213j
Joseph J, Jemmis E (2007) Red-, blue-, or no-shift in hydrogen bonds: a unified explanation. J Am Chem Soc 129:4620–4632. https://doi.org/10.1021/ja067545z
Liu SY, Dykstra CE (1986) A theory of vibrational transition frequency shifts due to hydrogen bonding. J Phys Chem 90:3097–3103. https://doi.org/10.1021/j100405a013
Matei I, Ionescu S, Hillebrand M (2010) Solute-solvent hydrogen bond formation in the excited state. experimental and theoretical evidence. In: Hydrogen Bonding and Transfer in the Excited State, vol 1. John Wiley and Sons pp 79–123. https://doi.org/10.1002/9780470669143.ch4
Acknowledgements
We gratefully acknowledge the Laboratório de Modelagem Molecular, GTI-CES and UFCG-CES for the help and support in this work. The authors thank Secretaria de Estado do Distrito Federal (SEEDF) and Fundação de Apoio e Pesquisa do Distrito Federal (FAPDF).
Funding
This work was supported by the Molecular Modeling Laboratory, GTI-CES, and Centro de Educação e Saúde of Universidade Federal de Campina Grande, CES-UFCG.
Author information
Authors and Affiliations
Contributions
AM Lima and GF Velardez performed the computer simulations and analysis of the results. AM Lima produced the figures under the supervision of NF Frazão and GF Velardez. AM Lima wrote the first draft of the manuscript. All the authors corrected, commented, and approved this version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
de Lima, A.M., Frazão, N.F. & Velardez, G.F. DFT and TD-DFT study of hydrogen bonded complexes of aspartic acid and n water (n = 1 and 2). J Mol Model 29, 94 (2023). https://doi.org/10.1007/s00894-023-05500-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-023-05500-z