
Abstract
Anthocyanidins, leucoanthocyanidins, and flavonols are natural compounds mainly known due to their reported biological activities, such as antiviral, antifungal, anti-inflammatory activities, and antioxidant activity. In the present study, we performed a comparative structural, conformational, electronic, and nuclear magnetic resonance analysis of the reactivity of the chemical structure of primary anthocyanidins, leucoanthocyanidins, and flavonoids. We focused our analysis on the following molecular questions: (i) differences in cyanidin catechols ( +)-catechin, leucocyanidin, and quercetin; (ii) the loss of hydroxyl presents in the R1 radical of leucoanthocyanidin in the functional groups linked to C4 (ring C); and (iii) the electron affinity of the 3-hydroxyl group (R7) in the flavonoids delphinidin, pelargonidin, cyanidin, quercetin, and kaempferol. We show unprecedented results for bond critical point (BCP) of leucopelargonidin and leucodelphirinidin. The BCP formed between hydroxyl hydrogen (R2) and ketone oxygen (R1) of kaempferol has the same degrees of covalence of quercetin. Kaempferol and quercetin exhibited localized electron densities between hydroxyl hydrogen (R2) and ketone oxygen (R1). Global molecular descriptors showed quercetin and leucocyanidin are the most reactive flavonoids in electrophilic reactions. Complementary, anthocyanidins are the most reactive in nucleophilic reactions, while the smallest gap occurs in delphinidin. Local descriptors indicate that anthocyanidins and flavonols are more prone to electrophilic attacks, while in leucoanthocyanidins, the most susceptible to attack are localized in the ring A. The ring C of anthocyanidins is more aromatic than the same found in flavonols and leucoanthocyanidins.
Methods
For the analysis of the molecular properties, we used the DFT to evaluate the formation of the covalent bonds and intermolecular forces. CAM-B3LYP functional with the def2TZV basis set was used for the geometry optimization. A broad analysis of quantum properties was performed using the assessment of the molecular electrostatic potential surface, electron localization function, Fukui functions, descriptors constructed from frontier orbitals, and nucleus independent chemical shift.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Flavonoids are natural compounds that can be obtained from several sources. These molecules have been reported with different biological activities, such as antibacterial, antiviral, antifungal, and anti-inflammatory activities [1,2,3,4]. The antioxidant activity has been the main biological activity reported for flavonoids, and it is involved in the prevention of the cell oxidative stress favoring the redox equilibrium [5]. Structures of anthocyanidins, leucoanthocyanidins, and flavonols have a remarkable distribution in plants and have in common broad health benefits, being applied for the design of new drugs [6,7,8]. Most recently, it was shown that quercetin and kaempferol flavonols have the potential to inhibit the 3CLpro protease of SARS-CoV-2 [8, 9].
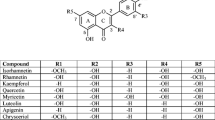
The primary flavonoids include anthocyanidins, leucoanthocyanidins, and flavonols [5]. Figure 1 shows the flavonoid scaffold composed by two phenyl rings A and B, and a heterocycle C, depicting the structure of nine primary flavonoids.

Molecular structure of nine primary flavonoids investigated in the present study
Hydroxyl and ketone groups determine the difference among these structures. R1 radicals are absent in anthocyanidins, while leucoanthocyanidins contain hydroxyl groups in R1, and flavonols contain ketones in R1 [5]. The anthocyanidins in the non-complexed form are found as cations, and these molecules perform an exciting function as pigmentation in plants [10]. Anthocyanidins and flavonols were widely investigated by experimental and computational methods. However, there is no sufficient information about the reactivity, aromaticity, and electronic properties of leucoanthocyanidins [10].
Leucoanthocyanidins are intermediate molecules obtained from the biosynthesis of anthocyanidins [11, 12]. They are found in converting the dihydroflavonoids in anthocyanidins in different species, such as Matthiola incana and Antirrhinum majus [11, 12]. Recently, studies have demonstrated that leucodelphinidin exhibits antiviral activity against the SARS-CoV-2. This compound has a high affinity against the Mpro and PLPro proteases from SARS-CoV2, [13], which induces conformational changes that impair their enzymatic activity [14].
Flavonoids may inhibit trypsin-like protease and are therapeutically valuable in treating tumors and cardiovascular disease [15]. Quercetin (C15H10O7) binds to the active sites of the urokinase-type plasminogen activator (uPA, PDB ID: 5XG4). B ring (catechol or benzenediol) binds via hydrogen bonding (S1 pocket) into the uPA, while two hydroxyl groups on ring A bind to the S2 pocket [15]. Catechol groups can inhibit most trypsin-like serine proteases.
Although phenolic compounds can be oxidized with rapid elimination through glucuronidation undertaken by detoxification enzymes, these compounds can also be synthetically modified to develop new drugs [15]. In addition, the comparative analysis of reactivity of quercetin could corroborate with the design of new bioactivity compounds.
The ( +)-catechin is a natural product and similarly to the cyanidin contains a hydrogen specie in R1 radical. Chloé Maugé et al. found a torsion angle equal to 76° between the rings C and B of the ( +)-catechin [16]. Other anthocyanidins show higher glycosylation activities, such as pelargonidin and cyanidin, presenting 29% and 17%, respectively. However, compared with other flavonols, such as quercetin and kaempferol, they show glycosylation activities of 5.4% and 4.5%, respectively [17]. Hiromoto et al. suggested that the difference in flavonoid activity against the enzyme must be due to substitutions in ring B [17].
The antioxidant and the reactivity of flavonoids against specific targets have relied on the bioactive hydroxyl groups in their structures [5]. Therefore, comparing these nine flavonoids (Fig. 1) can reveal contributions of kaempferol, quercetin, leucodelphinidin, and leucopelargonidin compared with the other five molecules.
Several quantum chemical descriptors have been widely used to characterize intramolecular and intermolecular interactions [18,19,20,21]. Conformational analysis is of interest in these flavonoids due to the large degree of freedom of these molecules, influenced mainly by hydrogen bonding interactions. Therefore, we have carried out a conformational analysis of the dihedral angle between the rings B and C. The aromaticity of these cyclic structures was described through the 1H chemical shift in the nuclear magnetic resonance (NMR) spectrum and is a key parameter to understand the reactivity [18]. The molecular electrostatic potential (MEP) surfaces were used for studying these molecular nucleophilic and electrophilic regions. Moreover, Fukui functions was used to study the molecular reactivity of compounds.
Computational details
Flavonoid molecular structures
The nine flavonoid molecules were selected searching three classes, anthocyanidins, leucoanthocyanidins, and flavonols: pelargonidin (PubChem CID 440,832, C15H11O5), cyanidin (PubChem CID 128,861, C15H11O6), ( +)-Catechin (PubChem CID 9064, C15H14O6); delphinidin (PubChem CID 128,853, C15H11O7), leucopelargonidin (PubChem CID 3,286,789, C15H14O6), leucocyanidin (PubChem CID 71,629, C15H14O7), leucodelphinidin (PubChem CID 3,081,374, C15H14O8), kaempferol (PubChem CID 5,280,863, C15H10O6), and quercetin (PubChem CID 5,280,343, C15H10O7).
Density functional theory calculations
All properties were studied using density functional theory (DFT) available in the Gaussian09 program in the gas phase (vacuum) [22]. The conformational and electronic properties were analyzed using the CAM-B3LYP [23] functional with the def2TZV basis set of Ahlrichs and coworkers assessed with polarized split-valence [24, 25]. For comparison analysis, we have added the Grimme D3 scheme for the van de Waals dispersion energy correction [26] and the solvent analysis using IEFPCM (water) method [27].
The conformational analysis in these flavonoids consists of variations in the dihedral angle between the benzopyran (heterocycle formed by the ring A and the pyran ring C) and the ring B. The conformational analysis was performed by varying the dihedral angle τ defined by the atoms O1–C2–C1′–C6′ with 18 steps of 10.0°. It is expected that significant changes in the dihedral angles occur due to weak interactions between hydroxyls of ring B and atoms of pyran ring C.
In addition, the topological analysis provides information on bond critical point (BCP) [28]. Thus, the degree of covalence of each BCP was determined through the AIMAII package [29]. The electronic properties consist of (i) analysis of the degree of covalence, (ii) BCP, (iii) electron localization function (ELF), and (iv) MEP isosurface analyses. These analyses were performed using the Multiwfn package [30]. Additionally, we used the visual molecular dynamic (VMD) program to visualize ELF isosurfaces [31]. MEP was plotted with electron density \(\rho =0.001 \mathrm{e}/{\mathrm{bohr}}^{3}\) for the van der Waals interactions. MEP is a useful tool to indicate nucleophilic and electrophilic regions in a molecule, helping to understand van der Waal and electrostatic interactions. In contrast, a useful tool to analyze the covalent bond interaction consists in the ELF analysis that points out regions with the most probable presence of electron pairs.
Fukui functions
Fukui functions are an alternative quantum chemistry analysis to study the molecular reactivity of compounds describing local electron densities and have been widely used to predict reactive sites of molecules [32,33,34,35]. The term that describes the removal of electrons from the molecule is represented by \({f}^{-}\) (electrophilic attack), while the term that describes the addition of electrons to the molecule is represented by \({f}^{+}\) (nucleophilic attack), according to Eqs. 1 and 2, respectively:
In addition to the local descriptors, global descriptors were analyzed, such as the electronic chemical potential (μ), chemical hardness (η), softness S, electrophilicity index ω, and electronegativity (χ). The study of the reactivity of polar molecules through Fukui functions was carried out analyzing the molecules as anions using the difference between the atomic charge of the anion and the neutral atom [36]. Based on Fukui functions, we also used this model to study the hybrid resonance structures in neutral, monocationic, and cationic molecules.
The procedure used in the Fukui functions of anthocyanidins was to shift \({N}_{0}\) to \({N}_{0}-1\) in the differences between the electronic energies of neutral systems and their cations (vertical ionization energy). The electrophilic and nucleophilic reactivities were calculated using the Fukui functions, using densities of \(\rho =0.007\) for \({f}_{{N}_{0}}^{-}\), \({f}_{{N}_{0}}^{+}\) and \(\rho =0.01\) for \({f}^{0}\) [30]. The HOMO (\({\varepsilon }_{H}\)) and LUMO (\({\varepsilon }_{L}\)) orbital energies are relevant to study the molecule reactivity. Larger values of \({\varepsilon }_{H}\) indicate greater reactivity in electrophilic reactions, while smaller \({\varepsilon }_{L}\) indicate nucleophilic reactions. The addition of unsaturation to aromatic rings can reduce the differences between \({\varepsilon }_{H-L}\) and thus increase the molecule reactivity.
NICS method
The hybrid HSEh1PBE functional with cc-pVDZ basis set was used to perform the nucleus-independent chemical shifts (NICS) method [37] under Gauge-independent atomic orbitals (GIAO) [38]. HSEh1PBE/cc-pVDZ was recently used to study the aromaticity of flavonoids [18]. NICS was calculated for the 1H-NMR with ghost atoms at the ring centerline and distances ranging from 1 up to 5 Å [37].
Results and discussions
A comparative analysis of the reactivity of different flavonoid classes was performed to shed light on the influence of hydroxyls in the functional groups attached to C4 (ring C).
Conformational analyses
Figure 2 shows energy barriers around 90.0° for anthocyanidins and flavonols, so these molecules are planar at the lowest energy conformations except for delphinidin. However, this behavior does not occur in leucoanthocyanidins, mainly due to the hydroxyl bonded to C4 (R1). A recent study using DFT showed that the O1–C2–C1′–C6′ dihedral angle (τ) of flavonols and anthocyanidins is planar at the lowest conformational energy. These structures show intramolecular bonds between the hydroxyls bonded to C3 and C5 carbons and the ketone group [39]. However, pelargonidin and delphinidin present an intramolecular bond between the O3 (C3 hydroxyl) and the C6′ [40, 41]. Cyanidin comprises the same planar dihedral angle (τ) using Hartree–Fock with an interaction between the O3 and C6′ [42].

Conformational analysis using CAM-B3LYP/def2TZV of the τ torsion angle (in degrees) of anthocyanidins, leucoanthocyanidins, and flavonols
Online Resource 1 shows the dihedral angles for the calculated molecules and the literature results. The calculated dihedral angles are τ (O1-C2-C1′-C6′), ω (C9-O1-C2-C3), and ϕ (C10-C4-C3-C2). Angles ω and ϕ define the torsion of the ring C. While angle τ represents the planarity of ring B concerning ring C, suggesting that this angle is representative of the whole planarity of the molecule.
Online Resource 1 shows the planar angles τ found in Fig. 2 for anthocyanidins and flavonols. The literature report for pelargonidin and delphinidin dihedral angles τ is equal to 13.60° and − 150.78°, respectively [40]. These values indicate that these molecules have distortion from planarity. Calculations show that leucoanthocyanidins are significantly twisted, in agreement with the reported results for leucocyanidin [43]. Different from the other cationic anthocyanidins, ( +)-catechin has a neutral charge. This significant difference reflects in the behavior of the dihedral angle. These molecules were also studied using Grimme D3 dispersion correction and water solvent effects. The results are in Online Resource 1. It is worthwhile to mention that these systems have a wide range of solvents [44]. Furthermore, studies in the literature show that there is a great difficulty in choosing a solvent to study the reactivity of these nine molecules presented in this work [44,45,46]. The results show that the structures changed only slightly with the largest RMSD of 0.281 Å under D3 dispersion and solvent calculations, corroborating the vacuum results. Therefore, the reactivity parameters studied in vacuum could be useful for studies in solvent.
Electronic structure analysis
Analyzing the BCP (3, − 1) critical points, it is possible to verify the interaction trends. If Hc < 0, then the interaction is expected to have a covalent component. If Hc > 0, then the interaction is non-covalent. Moreover, the degree of bonding can be classified through of Hc/ρc, which is the total energy per electron in the BCP point [47]. Figure 3 and Table 1 show the BCP (3, − 1), Hc, and Hc/ρc values for the investigated molecules. The intramolecular interactions (Fig. 3) are classified as type 1, between the hydroxyl hydrogen in R1 of ring C and oxygen in R2 of ring A. Type 2 is between hydrogen and hydroxyl in ring C, and type 3 is between hydrogen in the ring B and hydroxyl in the ring C.

Bond critical points (3, − 1) of the nine investigated molecules
Despite the pyrogallol group (ring B) is a known electrophile in the literature [48], quercetin pyrogallol moiety shows no influence on BCPs compared to kaempferol. The same was found for delphinidin compared to cyanidin. The results of Table 1 are in accordance to the literature for pelargonidin, delphinidin [40, 41], kaempferol, and quercetin [49].
Differently from the other molecules, kaempferol and quercetin show a value of Hc < 0 (covalent character) for the BCP (3, − 1) of type 1 interaction (Table 1), formed between the hydroxyl hydrogen (R2) and the oxygen of ketone (R1). The results of Hc/ρc show the same degree of covalent interaction of these two molecules. Positive Hc/ρc values for the other molecules indicate an electrostatic interaction.
ELF isosurfaces point out regions of highest probability for finding electrons. The ELF map presented in Fig. 4 is projected between 0.0 and 1.0, with the delocalized electrons in the range \(<0.5\) [50]. High electron localization is due to a covalent bond, a lone pair of electrons, or a nuclear charge [51].

ELF contour map of anthocyanidins and flavonols
The hydroxyl radicals in anthocyanidins and flavonols show localized electron densities, with the most localized densities found in cyanidin hydroxyls R4, R5, and R7. Localized electron densities between hydroxyl hydrogen (R2) and ketone oxygen (R1) are found in kaempferol and quercetin. Anthocyanidins have similar ELF patterns (Fig. 4) for R2, R3, R5, and R7 moieties. These hydroxyls have a different ELF profile than flavonols, showing the most localized red color related to the hydrogen bonding of R2 interacting with the carbonyl of R1.
Leucopelargonidin, leucocyanidin, and leucodelphinidin ELF isosurfaces are shown in Fig. 5. Ring A and ring B of these molecules follow the profile of anthocyanidins. However, ring C of leucopelargonidin is the most distorted of these molecules, and ring B of leucopelargonidin and leucodelphinidin showed highest localized densities. It is also possible to find localized densities in the hydroxyls of ring A of all leucoanthocyanidins. Flavonols, including the carbonyl group, tend to increase the hydrogen bonding regarding the R2 hydroxyl. At the same time, leucoanthocyanidins and anthocyanidins show similar profiles for the hydrogen bonding on R2 hydroxyl, corroborating BCP results.

ELF contour map of leucoanthocyanidins
The MEP was applied to analyze the reactive behavior of these molecules. Figure 6 shows MEP of anthocyanidins, leucoanthocyanidins, and flavonols. The red- and blue-colored regions of the potential areas are related to the regions susceptible to electrophilic and nucleophilic attacks, respectively [52].

MEP surfaces of anthocyanidins, leucoanthocyanidins, and flavonols. Dipole moment vectors are displayed in blue
Figure 6 shows three different groups of molecules, anthocyanidins, leucoanthocyanidins, and flavonols. The first group showed predominant contributions of the positive regions, the second group showed small contributions of negative regions, and the third group showed significant contributions of the negative regions. The pelargonidin, cyanidin, and delphinidin cations showed to be repulsive throughout all molecules, so they tend to be more prone to nucleophilic attacks. ( +)-Catechin is the only anthocyanidin prone to electrophilic attack. The ring C of flavonols showed repulsive potentials (positive charge densities), thus being susceptible to nucleophilic attacks. The same behavior was not verified in anthocyanidins and leucoanthocyanidins.
The dipole moment vectors indicate the regions where nucleophilic attacks are most likely to occur. Dipole moments are quite similar in leucoanthocyanidins, but the same similarity does not occur in flavonols. Although kaempferol and quercetin are flavonols, these results demonstrated that different reactivity is expected from them.
Table 2 shows that the most negative εHOMO occurs in cations (anthocyanidins) when compared with quercetin and leucocyanidin, suggesting that these molecules are more reactive in electrophilic reactions. The anthocyanidins have the smallest gap ΔεL-H; therefore, they are more reactive, and the smallest gaps occur in delphinidin. In addition, we found that the lowest chemical hardness, the highest electrophilicity index, and electronegativity occur in anthocyanidin cations.
Leucocyanidin has the highest dipole moment expected for leucoanthocyanidins due to the \(\tau\) dihedral values. High values of dipole moments can indicate high reactivity. Global descriptors and electronegativity also indicate that anthocyanidin is the more reactive molecule, but it is also necessary to verify reactivity through local descriptors. Fukui functions are shown in Figs. 7 and 8.

Fukui functions \({f}^{-}\) of the nine investigated molecules for sites favorable to electrophilic attacks

Fukui functions \({f}^{+}\) of the nine investigated molecules for sites favorable to nucleophilic attacks
The susceptibility to electrophilic attacks is directly related to the ability to donate electrons and is represented by the HOMO orbital. Figures 7 and 8 show positively charged regions suitable for electrostatic attraction and negative regions related to electrostatic repulsion. Anthocyanidin cations and flavonols are directed to the electrophilic attacks in the ϕ dihedral. In contrast, in leucoanthocyanidins, the most susceptible regions to the electrophilic attack are found in the A ring. The highest propensity for electrophilic ( +)-catechin attacks occur in the A ring. The catechol of both flavonols shows the same behavior. The oxygen of the R1 groups of leucoanthocyanidins and hydroxyl groups of anthocyanidins is not able to electrophilic attack.
Figure 8 suggests that flavonol catechol, leucoanthocyanin R1 hydroxyls, and anthocyanidin radicals are not suitable to nucleophilic attacks.
Magnetic analysis
Figure 9 shows the NICS values centered in ring A, B, and C. The NICS (1) values in the isotropic field are of interest to discuss the aromaticity. High negative values of the shielding NMR indicate diatropic currents (aromaticity) [18]. The ketone oxygen in the R1 radical can influence aromaticity due to the presence of π electrons associated with this atom [18]. Recently, a comparative study of aromaticity in anthocyanidins, leucoanthocyanidins, and flavonols clarified the importance of the ketone located at the R1 radical. In this study, the NICS values were calculated using HSEH1PBE/cc-pVDZ functional, which was shown suitable for flavonoid results [18].

NICS (ppm per distance r in Å) in the isotropic field of the nine molecules. Values in red refers to the ring C NICS
The NICS, ELF analysis, MEP, and gap support the planarity of ring C. The analysis of aromaticity using NICS corroborates the ELF analysis and shows that the ring C is distorted in the ELF isosurfaces of leucoanthocyanidins. The investigated flavonoids that present smaller gap values also present higher aromaticities regarding ring C. MEP surfaces of anthocyanidins, leucoanthocyanidins, and flavonols showed repulsive potentials in the ring C of kaempferol and quercetin. Leucoanthocyanidins present a large positive region in ring C, likewise the flavonols. In contrast, the NICS shows that higher aromaticity occurs in the ring C of flavonols more than in leucoanthocyanidins.
Conclusions
We have studied aromaticity, reactivity, conformational, and electronic properties of a set of flavonoids. The conformational analyses showed that anthocyanidins and flavonols have a planar conformation which agrees with the previous results reported in the literature. Different from the other studied molecules, leucoanthocyanidins are highly distorted and have hydroxyls at R1 radical. In the present study, we showed unprecedented results for leucopelargonidin and leucodelphirinidin.
BCPs (3, − 1) were found between the oxygen of the hydroxyl groups in R7 and the hydrogen of the O6′. Leucoanthocyanidins and flavonols have three BCP (3, − 1) types in radicals R7, R1, and R2. BCP (3, − 1) formed between the hydrogen of hydroxyl (R2) and the oxygen of ketone (R1) show a value of Hc < 0 for kaempferol and quercetin, with the same degrees of covalence in these three molecules. The hydroxyl radicals in anthocyanidins and flavonols showed localized electron densities. Kaempferol and quercetin presented localized electron densities between hydroxyl hydrogen (R2) and ketone oxygen (R1).
Global molecular descriptors showed quercetin and leucocyanidin are the most reactive flavonoids in electrophilic reactions. Complementary, anthocyanidins are the most reactive in nucleophilic reactions, while the smallest gap occurs in delphinidin. Anthocyanidins present the lowest hardness and the highest electrophilicity and electronegativity.
Local descriptors indicate that anthocyanidins and flavonols are more prone to electrophilic attacks in the ϕ dihedral, while in leucoanthocyanidins, the most attack-prone regions are in the ring A. The R5 radical is susceptible to nucleophilic attacks on all molecules. Leucocyanidin and leucodelphinidin present a considerable nucleophilic region in ring B. Local descriptors also indicate that flavonol, catechols, leucoanthocyanidin R1 hydroxyls, and anthocyanidin radicals are not susceptible to electrophilic and nucleophilic attacks. The ring C of anthocyanidins is more aromatic than the same found in flavonols and leucoanthocyanidins. The investigated flavonoids that present smaller gaps also present greater aromaticity.
Data availability
The datasets analyzed during the current study are included in this paper and its supplementary information file.
Code availability
N/A.
References
Srinivas K, King JW, Howard LR, Monrad JK (2010) Solubility and solution thermodynamic properties of quercetin and quercetin. J Food Eng 100:208–218. https://doi.org/10.1016/j.jfoodeng.2010.04.001
Zhang P, Mak JCW, Man RYK, Leung SWS (2019) Flavonoids reduces lipopolysaccharide-induced release of inflammatory mediators in human bronchial epithelial cells: structure-activity relationship. Eur J Pharmacol 865:172731. https://doi.org/10.1016/j.ejphar.2019.172731
Wang N, Zhou X, Cui B (2019) Synthesis of a polymeric imidazolium-embedded octadecyl ionic liquid-grafted silica sorbent for extraction of flavonoids. J Chromatogr A 1606:460376. https://doi.org/10.1016/j.chroma.2019.460376
Panche AN, Diwan AD, Chandra SR (2016) Flavonoids: an overview. J Nutr Sci 5:e47. https://doi.org/10.1017/jns.2016.41
He H-F, Wei K, Yin J, Ye Y (2020) Insight into tea flavonoids: composition and chemistry. Food Rev Int 37:812–823. https://doi.org/10.1080/87559129.2020.1721530
Pina F, Parola AJ, Melo MJ et al (2019) Chemistry of anthocyanins. In: Celli GB (ed) Brooks MS-L. Exploiting Targeted Delivery for Improved Health. Royal Society of Chemistry, Anthocyanins from Natural Sources, pp 34–76
Cherrak SA, Merzouk H, Mokhtari-Soulimane N (2020) Potential bioative glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: a molecular docking and simulation studies. PLoS One 15:e0240653. https://doi.org/10.1371/journal.pone.0240653
Rizzuti B, Grande F, Conforti F et al (2021) Rutin is a low micromolar inhibitor of SARS-CoV-2 main protease 3CLpro: implications for drug design of quercetin analogs. Biomedicines 9:375. https://doi.org/10.3390/biomedicines9040375
Jo S, Kim S, Shin DH, Kim M-S (2020) Inibition of SARS-CoV-3CL protease by flavonoids. J Enzyme Inhib Med Chem 35:145–151. https://doi.org/10.1080/14756366.2019.1690480
Khoo HE, Azlan A, Tang ST, Lim SM (2017) Anthocyanidins and anthocyanins: colored pigments as food, pharmaceutical ingredients, and the potential health benefits. Food Nutr Res 61:1361779. https://doi.org/10.1080/16546628.2017.1361779
Li H, Tian J, Yao Y-Y et al (2019) Identification of leucoanthocyanidin reductase and anthocyanidin reductase genes involved in proanthocyanidin biosynthesis in Malus crabapple plants. Plant Physiol Biochem 139:141–151. https://doi.org/10.1016/j.plaphy.2019.03.003
Heller W, Britsch L, Forkmann G, Grisebach H (1985) Leucoanthoeyanidins as intermediates in anthocyanidin biosynthesis in flowers of Matthiola incana R. Br Planta 163:191–196. https://doi.org/10.1007/BF00393505
Singh A, Mishra A (2021) Leucoefdin a potential inhibitor against SARS CoV-2 Mpro. J Biomol Struct Dyn 39:4427–4432. https://doi.org/10.1080/07391102.2020.1777903
Rudrapal M, Issahaku AR, Agoni C, et al (2021) In silico screening of phytopolyphenolics for the identification of bioactive compounds as novel protease inhibitors effective against SARS-CoV-2. J Biomol Struct Dyn 1–17. https://doi.org/10.1080/07391102.2021.1944909
Xue G, Gong L, Yuan C et al (2017) Structural mechanism of flavonoids in inhibiting serine proteases. Food Funct 8:2437–2443. https://doi.org/10.1039/C6FO01825D
Maugé C, Granier T, D’Estaintot BL et al (2010) Crystal structure and catalytic mechanism of leucoanthocyanidin reductase from Vitis vinifera. J Mol Biol 397:1079–1091. https://doi.org/10.1016/j.jmb.2010.02.002
Hiromoto T, Honjo E, Noda N et al (2015) Structural basis for acceptor-substrate recognition of UDP-glucose: anthocyanidin 3-O-glucosyltransferase from Clitoria ternatea. Protein Sci 24:395–407. https://doi.org/10.1002/pro.2630
de Souza Farias SA, da Costa KS, Martins JBL (2021) Analysis conformational, structural, magnetic, and eletronic properties related to antioxidant activity: Revisiting flavan, anthocyanidin, flavone, flavonol, isoflavone and flavan-3-ol. ACS Omega 6:8908–8918. https://doi.org/10.1021/acsomega.0c06156
Nascimento LA, Nascimento ÉCM, Martins JBL (2022) In silico study of tacrine and acetylcholine binding profile with human acetylcholinesterase: docking and electronic structure. J Mol Model 28:252. https://doi.org/10.1007/s00894-022-05252-2
Pereira WA, Nascimento ÉCM, Martins JBL (2021) Electronic and structural study of T315I mutated form in DFG-out conformation of BCR-ABL inhibitors. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2021.1935320
Rocha KML, Nascimento ÉCM, Martins JBL (2021) Investigation on the interaction behavior of afatinib, dasatinib, and imatinib docked to the BCR-ABL protein. J Mol Model 27:309. https://doi.org/10.1007/s00894-021-04925-8
Frisch MJ, Trucksr GW, Schlegel HB, et al (2009) Gaussian 09, Revision D.01. Gaussian
Yanai T, Tew DP, Handy NC (2004) A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem Phys Lett 393:51–57. https://doi.org/10.1016/j.cplett.2004.06.011
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys Chem Chem Phys 7:3297–3305. https://doi.org/10.1039/b508541a
Bakowies D, von Lilienfeld OA (2021) Density functional geometries and zero-point energies in ab initio thermochemical treatments of compounds with first-row atoms (H, C, N, O, F). J Chem Theory Comput 17:4872–4890. https://doi.org/10.1021/acs.jctc.1c00474
Grimme S, Ehrlich S, Goerigk L (2011) Effect of the damping function in dispersion corrected density functional theory. J Comput Chem 32:1456–1465. https://doi.org/10.1002/jcc.21759
Scalmani G, Frisch MJ (2010) Continuous surface charge polarizable continuum models of solvation. I. General formalism. J Chem Phys 132:114110. https://doi.org/10.1063/1.3359469
Wick CR, Clark T (2018) On bond-critical points in QTAIM and weak interactions. J Mol Model 24:142. https://doi.org/10.1007/s00894-018-3684-x
Keith TA (2017) AIMAll, TK Gristmill Software
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33:580–592. https://doi.org/10.1002/jcc.22885
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:27–28. https://doi.org/10.1016/0263-7855(96)00018-5
López P, Méndez F (2004) Fukui function as a descriptor of the imidazolium protonated cation resonance hybrid structure. Org Lett 6:1781–1783. https://doi.org/10.1021/ol049454h
de Almeida AL, Barbosa LPG, Santos RL, Martins JBL (2016) Chemical reactivity indices of the caffeine molecule. Rev Virtual Química 8:483–492. https://doi.org/10.5935/1984-6835.20160035
Cremer D, Kraka E (2010) From molecular vibrations to bonding, chemical reactions, and reaction mechanism. Curr Org Chem 14:1524–1560. https://doi.org/10.2174/138527210793563233
Fr B, Muthu S, Prasana JC et al (2018) Spectroscopic (FT-IR, FT-Raman) investigation, topology (ESP, ELF, LOL) analyses, charge transfer excitation and molecular docking (dengue, HCV) studies on ribavirin. Chem Data Collect 17–18:236–250. https://doi.org/10.1016/j.cdc.2018.09.003
Klyukin IN, Vlasova YS, Novikov AS et al (2021) Theoretical study of closo-borate anions [BnHn]2? (n = 5–12): bonding, atomic charges, and reactivity analysis. Symmetry (Basel) 13:464. https://doi.org/10.3390/sym13030464
Stanger A (2006) Nucleus-independent chemical shifts (NICS): distance dependence and revised criteria for aromaticity and antiaromaticity. J Org Chem 71:883–893. https://doi.org/10.1021/jo051746o
Cheeseman JR, Trucks GW, Keith TA, Frisch MJ (1996) A comparison of models for calculating nuclear magnetic resonance shielding tensors. J Chem Phys 104:5497–5509. https://doi.org/10.1063/1.471789
Michalík M, Biela M, Cagardová D, Lukes V (2020) Influence of catecholic ring torsion on hydroxyflavones. Acta Chim Slovaca 13:49–55. https://doi.org/10.2478/acs-2020-0008
Estévez L, Mosquera RA (2007) A density functional theory study on pelargonidin. J Phys Chem A 111:11100–11109. https://doi.org/10.1021/jp074941a
Estévez L, Mosquera RA (2008) Molecular structure and antioxidant properties of delphinidin. J Phys Chem A 112:10614–10623. https://doi.org/10.1021/jp8043237
Mihaiela A, Mihai M, Mariana Ş et al (2016) Theoretical determination of redox electrode pottential of cyanidin. J Serbian Chem Soc 81:177–186. https://doi.org/10.2298/JSC150715075A
Augustine C (2019) Unravelling the competence of leucocyanidin in free radical scavenging: a theoretical approach based on electronic structure calculations. J Struct Chem 60:198–209. https://doi.org/10.1134/S0022476619020045
Sillero L, Prado R, Welton T, Labidi J (2021) Energy and environmental analysis of flavonoids extraction from bark using alternative solvents. J Clean Prod 308:127286. https://doi.org/10.1016/j.jclepro.2021.127286
Exarchou V, Troganis A, Gerothanassis IP et al (2002) Do strong intramolecular hydrogen bonds persist in aqueous solution? Variable temperature gradient 1H, 1H–13C GE-HSQC and GE-HMBC NMR studies of flavonols and flavones in organic and aqueous mixtures. Tetrahedron 58:7423–7429. https://doi.org/10.1016/S0040-4020(02)00820-7
Álvarez-Diduk R, Ramírez-Silva MT, Galano A, Merkoçi A (2013) Deprotonation mechanism and acidity constants in aqueous solution of flavonols: a combined experimental and theoretical study. J Phys Chem B 117:12347–12359. https://doi.org/10.1021/jp4049617
Espinosa E, Alkorta I, Elguero J, Molins E (2002) From weak to strong interactions: a comprehensive analysis of the topological and energetic properties of the electron density distribution involving X-H⋯F–Y systems. J Chem Phys 117:5529–5542. https://doi.org/10.1063/1.1501133
Su H, Yao S, Zhao W et al (2021) Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. Nat Commun 12:3623. https://doi.org/10.1038/s41467-021-23751-3
Aparicio S (2010) A systematic computational study on flavonoids. Int J Mol Sci 11:2017–2038. https://doi.org/10.3390/ijms11052017
Arulaabaranam K, Mani G, Muthu S (2020) Computational assessment on wave function (ELF, LOL) analysis, molecular confirmation and molecular docking explores on 2-(5-Amino-2- Methylanilino)-4-(3-pyridyl) pyrimidine. Chem Data Collect 29:100525. https://doi.org/10.1016/j.cdc.2020.100525
Fr B, Prasana JC, Muthu S, Abraham CS (2019) Molecular docking studies, charge transfer excitation and wave function analyses (ESP, ELF, LOL) on valacyclovir : a potential antiviral drug. Comput Biol Chem 78:9–17. https://doi.org/10.1016/j.compbiolchem.2018.11.014
Murray JS, Politzer P (2011) The electrostatic potential: an overview. WIREs Comput Mol Sci 1:153–163. https://doi.org/10.1002/wcms.19
Funding
This work was supported by the Brazilian National Council for Scientific and Technological Development (CNPq 306682/2021–4) and the Foundation for Research of Federal District/Brazil (FAPDF 00193–00000229/2021–21).
Author information
Authors and Affiliations
Contributions
Sergio Antônio de Souza Farias: Conceptualization, Formal analysis, Investigation, Resources, calculations, writing — original draft. Kauê Santana da Costa: formal analysis, methodology, writing — review and editing. João B. L. Martins: formal analysis, methodology, writing — review and editing.
Corresponding author
Ethics declarations
Ethical approval
N/A
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This paper belongs to the Topical Collection IX Symposium on Electronic Structure and Molecular Dynamics – IX SeedMol
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
de Souza Farias, S.A., da Costa, K.S. & Martins, J.B.L. Comparative analysis of the reactivity of anthocyanidins, leucoanthocyanidins, and flavonols using a quantum chemistry approach. J Mol Model 29, 93 (2023). https://doi.org/10.1007/s00894-023-05468-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-023-05468-w