
Abstract
Nonlinear optical materials have been investigated recently due to their potential technological applications in information storage and communications. In this context, semi-organic crystals can effectively combine the desired nonlinear optical properties of amino acids with the promising mechanical and thermal properties of inorganic materials. In this work, we have synthesized and characterized a semi-organic crystal of the amino acid l-histidine and hydrofluoric acid and investigated the chemical interactions between the organic and inorganic moieties. The crystal of l-histidine bis(fluoride) has been produced by slow solvent evaporation and characterized by X-ray diffraction (XRD) crystallography and thermogravimetric and differential thermal analyses. The XRD conducted using the Rietveld method shows that the unit cell is orthorhombic with the P21212 space group and contains four l-histidine bis(fluoride) units. Both differential thermal analysis and temperature-dependent XRD show that the crystals are thermally stable up to 191°C and do not undergo phase transition. The computational Hirshfeld surface analysis of the crystal structure reveals the main intermolecular interactions. Density functional theory has been employed to calculate the ionic interaction energy and electrostatic potential maps and confirm the spontaneity of ionic association at 191°C. The combined experimental and computational results show that the thermal stability of the semi-organic l-histidine bis(fluoride) crystal makes it suitable for nonlinear optical applications in optical sensing and communication systems.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Second-order nonlinear optical materials (NLOs) have been the subject of several studies in recent decades due to their potential technological applications in optical communications, integrated optical systems, and information optical storage [1,2,3]. Currently, organic or inorganic optical crystals are being synthesized with a remarkable crystalline perfection [4]. However, each crystal class has certain advantages and disadvantages. On one hand, the purely organic crystals feature excellent NLO properties and a great birefringence, though they have a low thermal and mechanical stability [5]. On the other hand, inorganic materials have a high melting point, mechanical resistance, and chemical inertia; they have a low optical nonlinearity [6, 7]. In this context, new semi-organic compounds with properties of organic and inorganic crystals are being extensively explored, as they tend to combine the desired properties [8, 9]. In addition, a major advantage of most semi-organic materials is that upon forming ionic bonds, the resulting crystals acquire a higher probability of growth, greater thermal and mechanical resistance, and an increased chemical stability [6, 10,11,12,13].
Amino acids are bifunctional organic molecules that contain a carboxylic group (–COOH) and an amino group (–NH2). In α-amino acids, the characteristic side chain (R) of each amino acid is bonded to the same C atom (called alpha carbon, Cα) as the amino and carboxylic groups. As a result of being bonded to four different groups, the Cα of all α-amino acids except glycine is chiral [14, 15]. Thus, α-amino acids acquire active optical properties due to being chiral species and directing the crystallization to non-centrosymmetric spatial groups. Moreover, the dipolar nature of amino acids provides unique physical and chemical properties, making them promising candidates for NLO applications [16, 17]. That is, the presence of the –COO− and –NH3+ groups promotes the increase of the asymmetric polarizability of the organic material, providing asymmetry in the ground-state charge of the molecule, necessary for the development of second-order optical nonlinearity [12, 18,19,20].
Histidine is an essential α-amino acid, which can exist as two optical isomers, l-histidine and d-histidine. Only the l-isomer is bioactive. The l-histidine plays a vital role in several biological mechanisms, including the formation of hemoglobin and neurotransmitters in the brain and nervous system, and is essential for tissue growth and repair and glucose supply to the liver [21]. The l-histidine has a pKa close to neutrality and contains an imidazole side group [22,23,24,25].
The salts of l-histidine have attracted attention as promising materials for application in NLO after the verification that l-histidine tetrafluoroborate has better NLO properties than l-arginine phosphate monohydrate [19, 20, 26]. Since then, many researchers have synthesized and studied the properties of various crystals derived from this α-amino acid and proposed them as potential candidates for NLO applications [27, 28].
The l-histidine salt LHis·2HF has been synthesized for the first time in 1968 by Schmid using the reaction of l-histidine with hydrofluoric acid (HF) and water [29]. The synthesis of a semi-organic crystal of l-histidine hydrofluoric dihydrate and its NLO properties and the structure of a l-histidine bis(fluoride) crystal have also been reported [19, 26, 30]. The last salt contains delocalized π-electrons, electron donor, and acceptor groups in addition to fluoride ions strongly bonded to hydrogens in the organic moiety, conferring to its crystals high optical non-linearity and rapid optical response characteristics [30].
Electronic structure calculations have been widely used to study molecular systems and ionic associates with the aim of exploring their structural and thermodynamic properties [31]. Density functional theory (DFT) [32,33,34] is among the most extensively used model to explore NLO properties. For example, DFT has been used to investigate second-order NLO in l-histidine crystals, by determining the electrical dipole (μ), polarization (α), and hyperpolarization (β) of the crystal [35]. In addition, DFT has been employed to study chemical structure of crystals and for vibrational analysis [36,37,38,39,40,41].
A computational technique widely used to explore the intermolecular interactions between molecules in crystals is the Hirshfeld surface analysis [42]. Based on the description of atom-to-atom contacts, the Hirshfeld surface analysis provides an in-depth picture of how molecules come together in a crystalline structure [43, 44]. The Hirshfeld surface is a function of the sum of the electron density of atoms in a species divided by the sum of their closest neighbors, resulting in an isosurface that provides information about intermolecular interactions [45,46,47].
In this work, we synthesized l-histidine bis(fluoride) crystals by the slow evaporation method and characterized their structural and thermal properties by X-ray diffraction, thermogravimetric, and differential thermal analysis. The study is directed towards investigating the crystal’s structural and thermal stability at high temperatures, since these characterizations are important for the possible use of this material in NLO devices. In addition, computational studies using the DFT and Hirshfeld surface methods were conducted to understand the intermolecular interactions in the sample.
Materials and methods
LHis·2HF crystal synthesis
The crystals were synthesized by the slow evaporation method. Initially, a saturated aqueous solution of l-histidine (Sigma-Aldrich, purity ≤ 99%) and hydrofluoric acid (Impex, purity ≤ 48%) was prepared in a 1:2 mole ratio using deionized water. The solution was rotated on a shaker at 360 rpm for 2 h until complete homogenization and was then filtered through a filter paper with 25-μm pore. Subsequently, the solution was placed in a 50-mL beaker and covered with a plastic film, where several holes were drilled. Then, the covered beaker was placed in an oven with a controlled temperature of 35°C, favoring the slow evaporation of water and causing the supersaturation of the solution. The crystals were obtained after 37 days of storage.
Characterization
The X-ray diffraction (XRD) measurements were performed with the crystals pulverized using an Empyrean PANanalytical diffractometer aligned with Bragg-Brentano reflection geometry (θ-θ), using Cu Kα radiation (λ = 1.5418 Å), operating at a voltage of 40 kV, and a current of 30 mA. The patterns were obtained at a step size of 0.02° for 3 s, and 2θ interval range from 5° to 45°. The high-temperature XRD measurements were performed using an Anton-Paar TTK 450 chamber coupled with the diffractometer. The sample diffraction pattern was measured in the temperature range from 30 to 190 °C. Rietveld refinement using the GSAS-EXPGUI interface [48] was used to analyze the crystal XRD data. From the obtained values of the lattice parameters at various temperatures, it was possible to calculate the linear coefficients of thermal expansion for each crystallographic direction using the following linear functions:
where \({\alpha }_{\left[100\right]}\), \({\alpha }_{\left[010\right]}\), and \({\alpha }_{\left[001\right]}\) are the coefficients of thermal expansion; da, db, and dc are the variations of the lattice parameters, and dT is the temperature variation [49].
Thermal analysis
Thermal gravimetric analysis (TGA) and differential thermal analysis (DTA) were used to observe the variation in mass and differential thermal flow, respectively, of the sample when exposed to a controlled temperature increase, to identify events such as melting point, structural phase transition, and decomposition. The TGA and DTA measurements were carried out simultaneously in a Shimadzu DTG-60 thermogravimetric analyzer with an α-alumina open crucible in a nitrogen atmosphere at a flow rate of 100 mL∙min−1, in the temperature range from 30 to 500 °C, with a heating rate of 10 °C∙min−1.
DFT study
The DFT calculations were performed using the hybrid functional ωB97x-D with both dispersion and long-range interaction corrections, the 6-311++G(d,p) basis set, and the integral equation formalism version of the polarizable continuum model (IEF-PCM) of solvation, using water as the solvent (Ɛ = 78.4), as implemented in the Gaussian 16 software [50,51,52]. The DFT calculations include full geometry optimization, followed by vibrational analysis to confirm the optimized geometries as minima on the potential energy surface. The initial geometry of the main ionic associate containing 3 protonated histidine cations, connected by hydrogen bond to a fluoride anion, was taken from the XRD structure published by Petrosyan et al. [26]. This ionic associate was used as a representative block of the crystal. The charge and multiplicity for the whole associated system of [3LHisH2·F]5+ were defined as 5 and 1, respectively. Finally, the DFT results were analyzed with the Chemcraft software to visualize the optimized geometry and properties of the systems [53, 54].
The fluoride and protonated histidine ions were also studied individually. Then, variation of Gibbs free energy (ΔG), enthalpy (ΔH), entropy (ΔS), and total electronic energy corrected with ZPVE (ΔE) associated to the ionic association were calculated based on Eqs. 4, 5, 6, 7:
where \({G}_{T}\), \({G}_{H}\), and \({G}_{F}\) denoted the Gibbs free energies of the ionic associate, protonated histidine, and fluoride anion, respectively; \({H}_{T}\), \({H}_{H}\), and \({H}_{F}\) denoted the enthalpies of the ionic associate, protonated histidine, and fluoride anion, respectively; ET, EH, and EF were the total electronic energies corrected with the zero-point vibrational energy (ZVPE) of the ionic associate, protonated histidine, and fluoride anion, respectively. All thermodynamic quantities were calculated at 298.15 K and 464.15 K, both at 1 atm. Basis set superposition error (BSSE) was included in the calculation of the ionic association energies.
Hirshfeld surface
The Hirshfeld surfaces and two-dimensional (2D) fingerprint graphics were obtained using the Crystal Explorer 17 software [55], to enable a detailed analysis of the interactions between the chemical species in the crystal. The Hirshfeld surfaces were mapped as a function of normalized distance (dnorm), to provide three-dimensional (3D) representations of close contacts, defined in terms of distances from a given point on the surface to the nearest external (de) and internal (di) atom, and van der Waals radius (rvdW). The mapped Hirshfeld surfaces were rendered through a red-white-blue color scheme, where red was used for close contacts, white was used for contacts near the van der Waals radius, and blue was used for long distance contacts [45, 47]. The 2D graphs, given as functions of de and di, cover all intermolecular contacts, quantify specific interactions, and summarize the frequency of each combination [43, 44].
Results and discussion
X-ray diffractometry
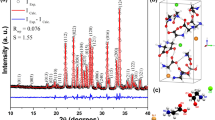
Figure 1(a) shows the Rietveld refinement of the LHis·2HF XRD pattern at room temperature. The Rietveld refinement R-factors for the weighted profile (RWP) and residual of least-squares refinement (RP) are 5.27% and 3.98%, respectively. The goodness of the fit indicator S is 1.65. These values indicate that the Rietveld refinement results obtained for the crystal have a high reliability [56].

a XRD Rietveld refinement of LHis·2HF crystal at room temperature; (b) unit cell of LHis·2HF; and (c) molecular structure of LHis·2HF, showing the hydrogen bonds as red sticks
The LHis·2HF at room temperature crystallizes in an orthorhombic phase (P21212-space group) with four molecules per unit cell (Fig. 1(b)) and has the following refined lattice parameters: a = 13,123(1) Å, b = 6.5810(0) Å, c = 9.651(5) Å, α = β = γ = 90°, and V = 833.499 Å3. These parameters are in agreement with those reported by Petrosyan et al. [26], with a percentage difference of less than 0.5%.
Figure 1(c) shows the LHis·2HF structure with its hydrogen bonds that are essential to maintain the material’s structural stability. According to Petrosyan et al. [26], LHis·2HF has a different hydrogen bonding scheme from LHis·2HCl and LHis·2HBr. There is a strong hydrogen bond between the acidic hydrogen atom of the carboxylic group of the α-amino acid and one of the fluoride ions: O2–H1···F1. Hydrogen bonds also form between the N–H proton of imidazole (when protonated) and the second fluoride ion: N2–H9···F2 and N3–H11···F2. When protonated (as –NH3+), the amino group also forms two hydrogen bonds with F1 and F2, namely N1–H3···F1, N1–H5···F1, and N1–H4···F2. Thus, each –NH3+ cation forms three hydrogen bonds.
Thermal analysis (TGA-DTA)
The LHis·2HF crystal thermogram is shown in Fig. 2. Analyzing the DTA curve, an endothermic peak is observed between 125 and 230 °C that was associated to the first weight loss (I) of approximately 20.70% in the TGA curve, equivalent to 0.861 mg of the initial crystal mass. This event is followed by two more stages of mass loss, i.e., (II) 231 to 320 °C, where an endothermic peak appears at 264 °C on the DTA curve, with a weight loss of 20.37% (0.847 mg) in the TG curve, and (III) 320 to 500 ºC, shows a weight loss of 14.43% (0.600 mg). These events are attributed to the decomposition of the LHis·2HF crystal.

TGA-DTA curves of the LHis·2HF crystal
From the TGA-DTA curve, it was observed that the LHis·2HF crystal is thermally stable up to 191 °C, without any transformation or transition in this temperature range (Fig. 2). Dhanuskodi and Ramajothi (2004) have performed a thermal and optical study of the crystal of l-histidine tetrafluoroborate [57] and also observed a good thermal stability up to its melting point of 235 °C. On the other hand, Madhavan et al. [19] have carried out DTA studies of the l-histidine hydrofluoride dihydrate crystal and revealed that the material showed thermal stability only up to 108 °C, due to the loss of lattice water. The value found by Madhavan et al. is lower than the one observed in our work. Thus, the good thermal stability of the LHis·2HF crystal can be attributed to the hydrogen bonding scheme, in conjunction with the anhydrous structure of the material, as rationalized by Faria et al. for LHis·HCl·H2O [24]. Thus, based on its thermal stability, LHis·2HF can be utilized for optoelectronic applications up to 191 °C.
XRD at high temperatures
The TGA-DTA results for LHis·2HF crystal show no evidence of the physical phenomenon of phase transition in the studied temperature range. To confirm the stability of the crystal, its thermal and structural behavior were also analyzed by using XRD at high temperatures. The crystal was subjected to XRD in the range from 30 to 190 °C, where the first decomposition event was detected by thermal analysis. The diffraction patterns are shown in Fig. 3.

Diffraction patterns of the LHis·2HF crystal as a function of temperature (a) 15° and 40°, (b) 17° and 18°; 18.5° and 20°; 26° and 27.5°; 31° and 32°
The diffraction patterns obtained between 30 and 190 °C show the LHis·2HF crystal only in an orthorhombic phase. In the diffractograms in Fig. 3(a), only small perceptible changes associated with the minimal structural changes in the crystal as a function of the temperature change are observed. In Fig. 3(b), it is possible to notice that some of the crystallographic axes undergo expansion or contraction with the sample’s gradual heating. The diffraction pattern undergoes a small displacement of the Bragg peaks to the left or to the right, accompanied by intensity loss at higher temperatures. This can be seen when following the dotted lines in Fig. 3(b) in the diffraction peaks with 2θ of 17.52°, 19.46°, 26.94°, and 31.60°.
After obtaining the lattice parameters as a function of the temperature, it was possible to estimate the crystal thermal expansion coefficients at temperature between 30 and 120 °C, using Eqs. 1, 2, and 3. Figure 4 shows the variation of the lattice parameters (ΔL/L0) as a function of the temperature, where the thermal expansion coefficient is calculated from the slope of each line, obtained as a result of a linear fit of all points. The calculated slopes are \(\alpha_{\left[100\right]}\) = –3.80(7) × 10–6 C−1, \(\alpha_{\left[010\right]}\) = 27.20(6) × 10–6 C−1, and \(\alpha_{\left[001\right]}\) = 7.13(3) × 10–6 C−1. These results show that the thermal expansion is negative along axis a and positive along axes b and c. Thus, the LHis·2HF crystal clearly exhibits anisotropic behavior, which may be related to the spatial orientation of the hydrogen bonds in the lattice, as reported for LHis·HBr·H2O [58].

Thermal expansion coefficient of the LHis·2HF crystal up to 100 °C.
DFT study
The geometries of the main ionic associate present in the crystal have been fully optimized by the means of DFT, using the ωB97x-D functional, 6-311++G(d, p) basis set, and IEF-PCM solvation method. The optimized geometries are confirmed as true minima on the potential energy surfaces by calculating the vibrational frequencies (confirmed by the presence of positive values only). Selected optimized structures are shown in Fig. 5(a). In the ionic associate, hydrogen bonds between the hydrogen atoms of the NH2 groups and the fluoride anion (F–) are observed in different regions of the protonated histidine. The results show that angles and interatomic distances converged to values close to those found experimentally in the crystal. Petrosyan et al. [26] reported that the N(imidazole)–F(fluoride) distances are 2.5421(16) Å and 2.5269(15) Å, and that the dihedral angle between the main chain carbon atoms of imidazole is −171.85(12)°. These values compare favorably with the optimized N(imidazole)–F(fluoride) distances of 2.5721 Å and 2.5685 Å and the imidazole dihedral angle of −179.09°.

a Optimized structure of the [3LHisH2·F]5+ associate; (b) experimental structure collected from the crystallographic database; (c) electrostatic potential of [3LHisH2·F]5+ mapped on the electron density surface; (d) electrostatic potential map
The analysis based on Eqs. 4–7 yields the ionic association Gibbs free energy, enthalpy, entropy, and electronic energy corrected with ZPVE values of −20.34 kcal∙mol−1, −52.59 kcal∙mol−1, −0.11 kcal∙mol−1∙K−1, and −52.98 kcal∙mol−1, respectively, at 298.15 K. After BSSE correction, the ionic association electronic energy corrected with ZPVE is −48.21 kcal∙mol−1. The values of Gibbs free energy, enthalpy, and electronic energy corrected with ZPVE and BSSE are negative. These parameters indicate that the formation of the ionic associate is exothermic and spontaneous, at room temperature. The spontaneous association is attributed to the formation of a network of strong hydrogen bonds between the protonated histidine and the fluoride anion [31, 59, 60].
The thermochemical analysis is also conducted at a temperature of 464.15 K. The calculated values of Gibbs free energy, enthalpy, and entropy are −2.63 kcal∙mol−1, −51.56 kcal∙mol−1, and 0.11 kcal∙mol−1∙K−1, respectively. When comparing the thermochemical results, it is observed that the Gibbs free energy undergoes a significant increase, as the temperature is increased from 298.15 to 464.15 K, but remains negative. The enthalpy and entropy undergo less significant changes. Our results also indicate that the increase in temperature favors an increase in the entropy of the system, which is related to an increase in the Gibbs free energy. The negative value of the Gibbs free energy of ionic association at 464.15 K (191°C) corroborate the experimental TGA-DTA findings that the material would be stable at this higher temperature.
The electrostatic potential of the ionic associate was mapped on the electron density isosurface (shown in Fig. 5(c)). The largest localization of negative charges, shown in red, are localized near the fluoride anion and the oxygen atoms of the carboxylate groups. The electrostatic potential map of the protonated l-histidine (Fig. 5(d)) shows the regions of positive electrostatic potential (in blue), which indicate favorable positions for nucleophilic attack, as well as regions with negative electrostatic potentials (in red), which indicate areas more favorable to electrophilic attack. In this case, the most positive potential is found near the –NH3+ groups, and the most negative potential near the carboxylic oxygen atoms.
Hirshfeld surface analysis
To better understand the intermolecular interactions in the LHis·2HF crystal, Hirshfeld surfaces were produced and analyzed with respect to the protonated histidine molecule, shown in Fig. 6(a). The strongest interactions can be seen in Fig. 6(b and c) as red regions, corresponding to the strongest hydrogen bonding sites. The interactions labeled as I, II, and VII correspond to the hydrogen bonds with F1 and F2, i.e., N1–H3···F1, N1–H5···F1 and N1–H4···F2 of the amino group, when protonated as –NH3+. Interactions IV and IX are those with the hydrogen bonds involving the imidazole N–H group through bonds N2–H9···F2 and N3–H11···F2 (atom labels are according to the notation of Fig. 1(c)). The hydrogen bond with the carboxylic group of the amino acid with one of the fluorine ions (O2–H1···F1) is shown in interaction VI. All the interactions mentioned agree with the XRD measurements, shown in Fig. 1(c). Interactions III, V, and VIII, indicated by white shading, are related to contacts with another protonated histidine in the neighborhood. When the crystalline lattice is expanded, a “beehive” structure is observed when seeing along the ab plane (Fig. 6(d)). When the crystal structure is rotated 90°, the orthorhombic structure is observed along the ac plane (see Fig. 6(e)). It is important to note that these Hirshfeld surfaces show intermolecular interactions that have not previously been reported in the literature.

a Structure of l-histidine protonated in the –NH3+ group; b Hirshfeld surface of protonated l-histidine mapped according to dnorm; c Hirshfeld surface rotated by 180º relative to a around the horizontal axis; d expanded LHis·2HF crystal structure along the a and b axes; and e expanded LHis·2HF crystal structure along the a and c axes
The 2D fingerprint plot provides a histogram as a function of the colored surface points fraction, in which the blue color represents few points and the red color represents many points, accounting for specific close contacts [43, 44, 47]. The calculation provides a quantitative information on the intermolecular interactions, which makes clearer the analysis of the interactions between the chemical species in the crystal.
The cumulative 2D fingerprint graph is shown in Fig. 7(a). The decomposed graphs of the H···F, H···H, H···O/O···H, H···C/C···H, and H···N/N···H interactions, with their respective percentage contributions, are shown in Fig. 7(b, c, d, e, and f) , respectively. The fingerprint graphs (Fig. 7(b–f)) correspond to the most important interactions, contributing to 93.3% to the total Hirshfeld surface. The remaining percentages are related to less significant interactions. In 2D fingerprint graphics, the presence of long, thin peaks in the regions of lower de + di values indicates the presence of strong interactions on the surfaces [61]. Long, thin peaks are seen in Fig. 7(b and d) , corresponding to the H···F and H···O/O···H interactions, confirming these as the strongest interactions in the crystal (Fig. 7(c)). Additionally, the H···H interaction is the most representative in terms of percentage contribution to interactions between atoms.

a Full 2D fingerprint graph of l-histidine protonated in –NH3+ and interaction-specific fingerprint graphs b H···F, c H···H, d H···O/O···H, e H···C/C···H and H···N/N···H
Figure 8 shows a more detailed model of the intermolecular contacts, showing the three most significant interactions of the analyzed Hirshfeld surfaces of the LHis·2HF crystal. The dashed orange, red, and green lines show the H···F, H···O/O···H, and H···H contacts, respectively. Thus, Fig. 8 corroborates the findings presented in Fig. 6(b and c) , showing that the main intermolecular interactions involve hydrogen bonding, confirming their substantial contributions to the stability of the crystal.

Intermolecular interactions of LHis·2HF crystal: dashed orange (H···F), red (H···O/O···H), and green lines (H···H)
Conclusions
In this work, LHis·2HF crystals were successfully synthesized by the slow evaporation technique and their thermal stability was investigated by using high-temperature XRD and thermal analysis with simultaneous TGA and DTA measurements. The TGA-DTA analyses showed that the l-histidine bis(fluoride) crystal was thermally stable up to the range of 191 °C. No events were observed in the DTA curve related to phase transitions, what was confirmed by the XRD analysis as a function of temperature. The effect of temperature on the crystallographic axes of the unit cell was verified in the orthorhombic phase, where a markedly anisotropic behavior was observed. These studies made it possible to estimate the thermal expansion coefficients of the crystal. The thermal stability of the crystal can be related to the hydrogen bonding and the lack of water in its matrix.
Moreover, DFT computational studies and Hirshfeld surface analysis were conducted to investigate the intermolecular hydrogen bonding interactions. In the DFT study, we obtained the optimized geometry of the main ionic associate in the crystal. The thermodynamic results confirmed the spontaneous formation of the ionic associate at temperature as high as 191°C, as a consequence of the occurrence of a strong attraction between the ions in this system, even in water. In the solid state, strong attraction between the ions can also be expected, which justifies the high stability of the solid.
The Hirshfeld surfaces identified the main points of intermolecular interactions in the structure. The Hirshfeld fingerprint graphs provided quantitative insights on the frequency of the occurrence of the different close contacts. The main points of intermolecular interactions match with the interactions indicated in the DFT study.
Taken together, the crystal characterization, thermal stability, and computational studies show that LHis·2HF crystal exhibits the thermal properties and possesses the intermolecular hydrogen bonding interactions that make it suitable for NLO applications. Also, these results contribute to developing a comprehensive understanding of the stabilization of semi-organic crystals with attractive NLO properties.
Funding
This research was supported by Fundação de Amparo à Pesquisa e ao Desenvolvimento Científico e Tecnológico do Maranhão (FAPEMA), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and the Program of Energy Research and Development (PERD).
Data availability
Data and materials are available on request from the authors.
Code availability
This work was conducted using the commercial software Gaussian 16, Chemcraft, and the freeware Crystal Explorer 17.
References
Sugandhi K, Verma S, Jose M et al (2013) Effect of pH on the growth, crystalline perfection, nonlinear optical and mechanical properties of tris-glycine zinc chloride single crystals. Opt Laser Technol 54:347–352. https://doi.org/10.1016/j.optlastec.2013.05.028
Suresh N, Selvapandiyan M, Mohammad F, Sagadevan S (2020) Influence of bismuth nitrate doping towards the characteristics of L-Alanine nonlinear optical crystals. Chinese J Phys 67:349–359. https://doi.org/10.1016/j.cjph.2020.07.014
Rajagopal Krishnamoorthy B, Kannan S, Sivaperuman K (2021) Synthesis and crystal growth of 2, 3-dimethyl-N-[2-(hydroxy) benzylidene] aniline: An organic second-order non-linear optical crystal for non-linear optical (NLO) applications. Opt Mater 113:110864. https://doi.org/10.1016/j.optmat.2021.110864
Palaninathan J, Rathinam V, Ali Baig AB et al (2020) Glycine doped L-alanine sodium sulphate single crystals for nonlinear optical applications. Mater Technol 00:1–10. https://doi.org/10.1080/10667857.2020.1859053
Senthilkumar M, Singh PK, Singh V et al (2020) Unidirectional seeded growth of l-glutamic acid hydrobromide single crystal and its characterization. Phase Transitions 93:83–90. https://doi.org/10.1080/01411594.2019.1670831
Kannan V, Ganesh RB, Ramasamy P (2006) Growth of new nonlinear optical crystals of hydrochlorides of L-histidine from solution. Cryst Growth Des 6:1876–1880. https://doi.org/10.1021/cg0601960
Rathee SP, Ahlawat DS, Dhas SAMB et al (2020) Investigations on key aspects of solution growth L-alanine strontium chloride trihydrate single crystal for non-linear optical and photonic applications. Solid State Commun 319:114010. https://doi.org/10.1016/j.ssc.2020.114010
Nalini H, Thairiyaraja M, Arunagiri C et al (2021) Crystal growth, structural, nonlinear, optical and theoretical investigations of L-histidinium trichloroacetate single crystals. Bull Mater Sci 44:1–17. https://doi.org/10.1007/s12034-020-02319-9
John JS, Saravana Kumar M, Joy LK et al (2020) Crystal growth, dielectric studies, charge transfer and ionic hydrogen-bonding interactions of L-arginine hydrobromide monohydrate single crystal: a novel third order nonlinear optical material for optoelectronic applications. Opt Laser Technol 125:106043. https://doi.org/10.1016/j.optlastec.2019.106043
Aggarwal MD, Stephens J, Batra AK, Lal RB (2003) Bulk crystal growth and characterization of semiorganic nonlinear optical materials. J Optoelectron Adv Mater 5:555–562
Kirubavathi K, Selvaraju K, Valluvan R et al (2008) Synthesis, growth, structural, spectroscopic and optical studies of a new semiorganic nonlinear optical crystal: l-Valine hydrochloride. Spectrochim Acta - Part A Mol Biomol Spectrosc 69:1283–1286. https://doi.org/10.1016/j.saa.2007.07.042
Praveen Kumar P, Manivannan V, Tamilselvan S et al (2008) Growth and characterization of a pure and doped nonlinear optical l-histidine acetate single crystals. Opt Commun 281:2989–2995. https://doi.org/10.1016/j.optcom.2008.01.058
Zhang Y, Li H, Xi B et al (2008) Growth and characterization of l-histidine nitrate single crystal, a promising semiorganic NLO material. Mater Chem Phys 108:192–195. https://doi.org/10.1016/j.matchemphys.2007.09.006
Morimoto H, Unoura K, Nabika H (2021) Effect of amino acid addition on the crystallization of asparagine. Cryst Growth Des 21:2205–2211. https://doi.org/10.1021/acs.cgd.0c01653
Nugrahani I, Jessica MA (2021) Amino acids as the potential co-former for co-crystal development: a review. Molecules 26:3279. https://doi.org/10.3390/molecules26113279
Palaninathan J, Ali Baig AB, Rathinam V et al (2021) Studies on the structural, thermal, optical and nonlinear optical characteristics of a novel organic glycine doped l-arginine adipate crystals. J Mater Sci Mater Electron 32:6820–6829. https://doi.org/10.1007/s10854-021-05387-7
Aishwarya P, Ilango E, Ramalingam G, Vetrivelan V (2021) Crystal growth, structural, optical, thermal, dielectric, mechanical and NLO studies of L-tyrosine zinc carbonate single crystals. Mater Today Proc 49:2574–2579. https://doi.org/10.1016/j.matpr.2021.05.556
Harvey RA, Ferrier DR (2011) Biochemistry. Lippincott Williams & Wilkins, Philadelphia, PA, USA
Madhavan J, Aruna S, Prabha K et al (2006) Growth and characterization of a novel NLO crystal l-histidine hydrofluoride dihydrate (LHHF). J Cryst Growth 293:409–414. https://doi.org/10.1016/j.jcrysgro.2006.05.050
Madhavan J, Aruna S, Thomas PC et al (2007) Growth and characterization of L-histidine hydrochloride monohydrate single crystals. Cryst Res Technol 42:59–64. https://doi.org/10.1002/crat.200610771
Holeček M (2020) Influence of histidine administration on ammonia and amino acid metabolism: A review. Physiol Res 69:555–564. https://doi.org/10.33549/physiolres.934449
de Menezes AS, dos Santos AO, Almeida JMA et al (2007) Piezoelectric coefficients of L-histidine hydrochloride monohydrate obtained by synchrotron x-ray Renninger scanning. J Phys Condens Matter 19:106218. https://doi.org/10.1088/0953-8984/19/10/106218
Remédios CMR, Paraguassu W, Lima JA et al (2008) Effect of Ni(II) doping on the structure of L-histidine hydrochloride monohydrate crystals. J Phys Condens Matter 20:1–6. https://doi.org/10.1088/0953-8984/20/27/275209
Faria JLB, Almeida FM, Pilla O et al (2004) Raman spectra of L-histidine hydrochloride monohydrate crystal. J Raman Spectrosc 35:242–248. https://doi.org/10.1002/jrs.1142
Fleck M, Petrosyan AM (2014) Salts of amino acids. Springer Cham, London, UK
Petrosyan HA, Karapetyan HA, Antipin MY, Petrosyan AM (2005) Nonlinear optical crystals of l-histidine salts. J Cryst Growth 275:1919–1925. https://doi.org/10.1016/j.jcrysgro.2004.11.258
John NL, Abraham S, Sajan D et al (2020) Molecular structure, NLO properties and vibrational analysis of L-histidine tetra fluro borate by experimental and computational spectroscopic techniques. Spectrochim Acta - Part A Mol Biomol Spectrosc 226:117615. https://doi.org/10.1016/j.saa.2019.117615
Yadav S, Kumari M, Nayak D et al (2020) Growth and characterization of single crystals of l-histidine hydrochloride monohydrate for nonlinear optical applications. J Electron Mater 49:7502–7508. https://doi.org/10.1007/s11664-020-08526-8
Muttenz HS (1968) Addition compounds of amino acids and hydrofluoric acid or soluble fluorides and method of preparing the same. United States Patent Office - US3413326A
Ramos Silva MR, Matos Beja A, Paixão JA (2004) Very short O-H⋯hydrogen bond in L-histidinium difluoride. J Fluor Chem 125:695–699. https://doi.org/10.1016/j.jfluchem.2003.11.020
Moniruzzaman (2019) Thermodynamic, HOMO-LUMO, MEP and ADMET studies of metronidazole and its modified derivatives based on DFT. Open Acess J Biomed Eng Biosci 3:262–266. https://doi.org/10.32474/OAJBEB.2019.03.000153
Chermette H (1998) Density functional theory a powerful tool for theoretical studies in coordination chemistry. Coord Chem Rev 178–180:699–721. https://doi.org/10.1016/s0010-8545(98)00179-9
Chermette H (1999) Chemical reactivity indexes in density functional theory. J Comput Chem 20:129–154. https://doi.org/10.1002/(SICI)1096-987X(19990115)20:1%3c129::AID-JCC13%3e3.0.CO;2-A
Hammud HH, Kortz U, Bhattacharya S et al (2020) Structure, DFT studies, magnetism and biological activity of bis[(µ-azido)-chloro-(1,10-phenanthroline)-copper(II)] complex. Inorganica Chim Acta 506:119533. https://doi.org/10.1016/j.ica.2020.119533
Novelli G, Maynard-Casely HE, McIntyre GJ et al (2020) Effect of high pressure on the crystal structures of polymorphs of l -histidine. Cryst Growth Des 20:7788–7804. https://doi.org/10.1021/acs.cgd.0c01085
Ben Ahmed A, Feki H, Abid Y et al (2008) Structural, vibrational and theoretical studies of l-histidine bromide. J Mol Struct 888:180–186. https://doi.org/10.1016/j.molstruc.2007.11.056
Ben Ahmed A, Feki H, Abid Y et al (2009) Crystal structure, vibrational spectra and theoretical studies of l-histidinium dihydrogen phosphate-phosphoric acid. J Mol Struct 920:1–7. https://doi.org/10.1016/j.molstruc.2008.09.029
Ben Ahmed A, Feki H, Abid Y et al (2010) Crystal studies, vibrational spectra and non-linear optical properties of l-histidine chloride monohydrate. Spectrochim Acta - Part A Mol Biomol Spectrosc 75:293–298. https://doi.org/10.1016/j.saa.2009.10.026
Maia JR, Lima JA, Freire PTC et al (2013) FT-IR and Raman spectra and DFT calculations on bis(L-histidinato) nickel(II) monohydrate. J Mol Struct 1054–1055:143–149. https://doi.org/10.1016/j.molstruc.2013.09.042
Sun GH, Sun XT, Sun ZH et al (2009) Growth and characterization of a nonlinear optical crystal: l-histidine trifluoroacetate. J Cryst Growth 311:3904–3910. https://doi.org/10.1016/j.jcrysgro.2009.06.022
Sethuram M, Dhandapani M, Sethu Raman M et al (2014) Structure and spectral properties of l-histidinium dipicrate dihydrate. Spectrochim Acta - Part A Mol Biomol Spectrosc 118:102–111. https://doi.org/10.1016/j.saa.2013.08.080
Li S, Bu R, Gou RJ, Zhang C (2021) Hirshfeld surface method and its application in energetic crystals. Cryst Growth Des 21:6619–6634. https://doi.org/10.1021/acs.cgd.1c00961
Tan SL, Jotani MM, Tiekink ERT (2019) Utilizing Hirshfeld surface calculations, non-covalent interaction (NCI) plots and the calculation of interaction energies in the analysis of molecular packing. Acta Crystallogr Sect E Crystallogr Commun 75:308–318. https://doi.org/10.1107/S2056989019001129
Spackman MA, Jayatilaka D (2009) Hirshfeld surface analysis. CrystEngComm 11:19–32. https://doi.org/10.1039/b818330a
Matos CRMO, Vitorino LS, De Oliveira PHR et al (2016) Supramolecular assembly of (Z)-ethyl 2-cyano-3-((4-fluorophenyl)amino) acrylate, crystal structure, Hirshfeld surface analysis and DFT studies. J Mol Struct 1120:333–340. https://doi.org/10.1016/j.molstruc.2016.05.033
Wang H, Xiao H, Liu N et al (2015) Three new compounds derived from nitrofurantoin: X-ray structures and Hirshfeld surface analyses. Open J Inorg Chem 5:63–73. https://doi.org/10.4236/ojic.2015.53008
McKinnon JJ, Jayatilaka D, Spackman MA (2007) Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem Commun 00:3814–3816. https://doi.org/10.1039/b704980c
Toby BH (2001) EXPGUI, a graphical user interface for GSAS. J Appl Crystallogr 34:210–213. https://doi.org/10.1107/S0021889801002242
Santos AO, Lang R, Sasaki JM, Cardoso LP (2019) Assessment of phase transition and thermal expansion coefficients by means of secondary multiple reflections of Renninger scans. J Appl Crystallogr 52:1271–1279. https://doi.org/10.1107/s1600576719011944
Da Chai J, Head-Gordon M (2008) Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys Chem Chem Phys 10:6615–6620. https://doi.org/10.1039/b810189b
Lage MR, Morbec JM, Santos MH et al (2017) Natural polyprenylated benzophenone: keto-enol tautomerism from density functional calculations and the AIM theory. J Mol Model 23:1–7. https://doi.org/10.1007/s00894-017-3251-x
Tomasi J, Mennucci B, Cammi R (2005) Quantum mechanical continuum solvation models. Chem Rev 105:2999–3093. https://doi.org/10.1021/cr9904009
Frisch MJ, Trucks GW, Schlegel HB et al (2016) Gaussian 16, Revision C.01, Gaussian, Inc. Wallingford, CT, USA
Chemcraft (2020) Graphical software for visualization of quantum chemistry computations. https://www.chemcraftprog.com
Spackman PR, Turner Michael J, McKinnon JJ et al (2021) Crystal Explorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J Appl Cryst 54:1006–1011. https://doi.org/10.1107/S1600576721002910
Young RA (1993) The rietveld method. Oxford University Press, New York, NY, USA.
Dhanuskodi S, Ramajothi J (2004) Crystal growth, thermal and optical studies of L-histidine tetrafluoroborate: a semiorganic NLO material. Cryst Res Technol 39:592–597. https://doi.org/10.1002/crat.200310229
Moura GM, Carvalho JO, Silva MCD et al (2015) Structural, vibrational and thermal characterization of phase transformation in l-histidinium bromide monohydrate single crystals. Mater Chem Phys 165:150–155. https://doi.org/10.1016/j.matchemphys.2015.09.009
Frimpong E, Skelton AA, Honarparvar B (2017) DFT study of the interaction between DOTA chelator and competitive alkali metal ions. J Mol Graph Model 76:70–76. https://doi.org/10.1016/j.jmgm.2017.06.025
Melengate GS, Quattrociocchi DGS, SiqueiraJúnior JM et al (2019) DFT study of the interaction between the Ni2+ and Zn2+ metal cations and the 1,2-dithiolene ligands: Electronic, geometric and energetic analysis. J Braz Chem Soc 30:1161–1177. https://doi.org/10.21577/0103-5053.20190011
Al-Wahaibi LH, Joubert J, Blacque O et al (2019) Crystal structure, Hirshfeld surface analysis and DFT studies of 5-(adamantan-1-yl)-3-[(4-chlorobenzyl)sulfanyl]-4-methyl-4H-1,2,4-triazole, a potential 11β-HSD1 inhibitor. Sci Rep 9:1–11. https://doi.org/10.1038/s41598-019-56331-z
Acknowledgements
The authors thank the Fundação de Amparo à Pesquisa e ao Desenvolvimento Científico e Tecnológico do Maranhão (FAPEMA), the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for the financial support. S.R.S. acknowledges support from the Government of Canada’s Program of Energy Research and Development.
Funding
Open Access provided by Natural Resources Canada.
Author information
Authors and Affiliations
Contributions
Ian Felipe Sousa Reis: material preparation, data collection, analysis, and writing—original draft preparation
Jailton Romão Viana: simulations, data collection, analysis, and writing—original draft preparation
João Gomes de Oliveira Neto: material preparation, data collection, analysis, and writing—original draft preparation
Stanislav R. Stoyanov: reviewing and editing, resources, and formal analysis
José Walkimar de M. Carneiro: reviewing and editing, resources, and formal analysis
Mateus Ribeiro Lage: conceptualization, reviewing and editing, resources, and supervision
Adenilson Oliveira dos Santos: conceptualization, reviewing and editing, resources, and supervision
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Reis, I.F.S., Viana, J.R., de Oliveira Neto, J.G. et al. Synthesis, characterization, and thermal and computational investigations of the l-histidine bis(fluoride) crystal. J Mol Model 28, 222 (2022). https://doi.org/10.1007/s00894-022-05168-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-022-05168-x