
Abstract
COVID-19 has recently grown to be pandemic all around the world. Therefore, efforts to find effective drugs for the treatment of COVID-19 are needed to improve humans’ life quality and survival. Since the main protease (Mpro) of SARS-CoV-2 plays a crucial role in viral replication and transcription, the inhibition of this enzyme could be a promising and challenging therapeutic target to fight COVID-19. The present study aims to identify alkaloid compounds as new potential inhibitors for SARS-CoV-2 Mpro by the hybrid modeling analyses. The docking-based virtual screening method assessed a collection of alkaloids extracted from over 500 medicinal plants and sponges. In order to validate the docking process, classical molecular dynamic simulations were applied on selected ligands, and the calculation of binding free energy was performed. Based on the proper interactions with the active site of the SARS-CoV-2 Mpro, low binding energy, few side effects, and the availability in the medicinal market, two indole alkaloids were found to be potential lead compounds that may serve as therapeutic options to treat COVID-19. This study paves the way for developing natural alkaloids as stronger potent antiviral agents against the SARS-CoV-2.
Similar content being viewed by others

Introduction
The coronavirus disease (COVID-19) spread to all countries and caused over four million deaths worldwide and continues to evolve. Therefore, the COVID-19 outbreak has emerged as a substantial and urgent risk to global health. Several clinical trials are in progress to alleviate the symptoms of COVID-19 patients, such as dry cough, dyspnea, and respiratory syndrome. To date, the combination of lopinavir-ritonavir (anti-HIV protease), nelfinavir (anti-HIV protease), favipiravir (anti-influenza), their families as a protease inhibitor, remdesivir as the inhibitor of RNA-dependent RNA-polymerases (RdRp), and chloroquine (anti-malaria) have been prescribed for patients with the covid-19 disease [1]. However, their clinical efficacy, mechanism of action, and severe side effects need further evaluation. To date, there are no FDA-approved drugs targeting COVID-19, and an effective approach to identify novel lead compounds is required.
Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), causing the COVID-19 disease, is categorized as beta-coronaviruses. Coronaviruses are a class of viruses belonging to the Coronaviridae (CoV) family. However, their mortality rate, pathogenesis, and transmission modes are different [2]. SARS-CoV-2, like other severe acute respiratory syndrome-related coronaviruses, is a spherical and positive-sense single-stranded RNA (+ ssRNA) virus. SARS-CoV-2 enters the human cells by binding to the cell membrane ACE2 receptor, replicating its genome, and subsequently translating inside the host cells [3]. Viral RNAs are translated into polypeptides that include several proteins [4]. Proteinase plays a vital role in recognizing and breaking the viral polypeptides into functional enzymes essential for the assembly and survival of viruses [5]. Therefore, viral protease inhibitors could be considered a potential agent to combat viral diseases like COVID-19.
Therapeutic targets in SARS-CoV-2 are shown in Fig. 1. The main protease (Mpro) of SARS-CoV-2 has been considered a promising therapeutic target for COVID-19 due to its crucial role in viral replication and transcription [6]. However, the suggested inhibitors of SARS-CoV-2 Mpro have not been successful for the therapeutic usage against COVID-19. Thus, efforts for finding new inhibitors of SARS-CoV-2 Mpro, with improved efficacy and low toxicity, are indispensable for managing the disease.

The overview diagram of therapeutic targets in SARS-CoV-2
Thousands of plants and their extracts are used as a source of medicine. Plant-based products play a substantial role in drug development and the treatment of several disorders. Several compounds have been reported to exhibit antiviral bioactivity against influenza virus, dengue virus, enterovirus, coronavirus, HIV, and hepatitis B [7, 8]. Some of these compounds have alkaloid structures [9, 10]. Structurally, the term alkaloid refers to a cyclic organic compound containing at least one nitrogen atom. Consequently, they can also be a source of new inhibitors for replicating SARS-CoV-2 Mpro. Despite the clear advantages of natural products in drug discovery, this source has rarely been applied to treat COVID-19.
There are various methods in drug discovery; however, computer-aided drug design (CADD) as a systematic and economical tool shows a high potential to recognize novel lead compounds [11], since the drug discovery and development, from identifying potential lead compounds to the approved medications, are an expensive and lengthy process. At the same time, combined computational approaches could assist as a promising opportunity for figuring out potential drug candidates from compound databases [6]. Accordingly, CADD analyses have been widely used to find anti-COVID-19 compounds through binding with the SARS-CoV-2 Mpro [12, 13].
This effort aims to use natural alkaloids for seeking potential therapeutic agents against COVID-19. For this purpose, docking-based virtual screening was carried out on a selected library of natural alkaloids and some of the antiviral FDA-approved drugs against the active site of SARS-CoV-2 Mpro. Afterward, molecular dynamic (MD) simulations were performed to validate the docking results and assess the stability of the best ligands in a dynamic environment. Eventually, the binding energy was calculated using the molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) approach. The obtained binding energies were applied to determine the binding affinity of compounds with SARS-CoV-2 Mpro. The potential inhibitors of SARS-CoV-2 identified through this analysis could be therapeutic options for drug discovery to combat the coronavirus outbreak.
Methods
Molecular docking studies
In order to characterize the binding energy and binding modes of natural alkaloids in the active site of SARS-CoV-2 Mpro and provide initial topology and coordinates parameters for the MD simulations, molecular docking analyses were performed using the AutoDock 4.2 software [14]. The crystal structure of SARS-CoV-2 Mpro (PDB code: 6LU7), which was recently issued, was obtained from the Protein Data Bank (www.rcsb.org). All input files and analyzing the results were prepared using AutoDockTools (ADT; Ver.1.5.6). PDBQT format of the protein was prepared by omitting the water molecules and other heteroatoms, including ligands and ions, from the PDB file. After adding polar hydrogen atoms, the partial atomic charges for all atoms were computed using the Kollman-united charges method. A list of natural alkaloids used to manage different diseases was compiled based on the literature review [15,16,17]. The 3D structures of natural alkaloids were achieved from the PubChem database in the structure-data file (SDF) format. Also, we examined some FDA-approved antiviral drugs that are newly introduced as potential inhibitors of SARS-CoV-2, including favipiravir, lopinavir, nelfinavir, ritonavir, and remdesivir [18].
Open Babel (version 2.3.1) [19] was used to convert SDF to PDB format. Atomic charges of all ligands were defined based on the Gasteiger-Marsili charge and saved in PDBQT format for the docking process by AutoDock 4.2. According to the most crucial reported residues, the grid box of 50 × 50 × 50 Å (x, y, and z) was specified on the protein binding pocket with a grid spacing of 0.375 Å for each dimension. All grid maps were produced by AutodGrid 4.2 [14]. Each docking round was carried out in 100 runs. Docking validation was performed using the extraction of the irreversible inhibitor N3 (PDB ID 6LU7) from the crystal structure and re-docking against its cognate receptor. Also, the validation of the procedure was examined via molecular docking study of PF-00835231, as a known SARS-CoV-2 Mpro inhibitor [20]. Eventually, the obtained results were ranked following the lowest binding energy and then analyzed to characterize the binding modes of the ligands in the protein’s binding pocket. ADT, Discovery Studio visualizer (Ver. 17.2) [21], and PyMOL Tcl [22] were used to visualize the obtained results from docking studies.
Molecular dynamic simulations
In order to assess the binding interactions of selected compounds with SARS-CoV-2 Mpro, MD simulations were run on a high-performance Linux cluster via the GROMACS 2020.1 package [23]. MD simulations have been carried out based on our previous work using the AMBER99SB-ILDN force field [11]. The protein–ligand complexes extracted from the docking analysis were used for MD simulations. The simulations were conducted on three individual systems, including the bare SARS-CoV-2 Mpro, SARS-CoV-2 Mpro- yohimbine, and SARS-CoV-2 Mpro-ajmalicine during 150 ns. All of the topology files of ligands were generated by the ACPYPE program package [24]. Initially, to prepare the input file for MD simulations, the best ligand–protein complex obtained from docking studies was chosen based on the lowest docking score and convenient interaction of ligands in the active site. The PROPKA 3.1 webserver was utilized to determine the protonation states of SARS-CoV-2 Mpro residues [25]. During the MD simulation, the protein–ligand complex was dunked in a dodecahedron shape box with a simple point charge (SPC) water model. Na+ and Cl− as opposite ions were used to neutralize the net charge of the system. The entire system was energy-minimized via 10,000 steepest descents steps. To equilibrate the system at a constant temperature of 300 K, NVT ensemble MD simulation was carried out by a 400 ps MD run. Then, an NPT ensemble was performed at a time duration of 1 ns in a periodic boundary condition. Temperature and pressure were kept constant employing Berendsen barostat and thermostat at 300 K and 1 bar with a coupling time of τT = 0.1 ps, τp = 0.5 ps, respectively. The electrostatic interactions and bond lengths were estimated via the Particle Mesh Ewald (PME) and the LINCS algorithm. Eventually, the system was subjected to MD simulations run for 150 ns in triplicate.
Calculation of binding free energy
The binding energy of ajmalicine and yohimbine, as the best compounds for the inhibition of SARS-CoV-2 Mpro and the binding energy contribution of residues, was computed based on the MM-PBSA method by g_mmpbsa v2020.1 package [26]. A detailed description of the method is provided in the previous study [27].
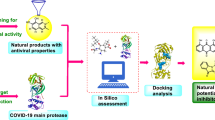
The workflow of the procedures used in the present study is depicted in Fig. 2.

Workflow of the methodology used for virtual screening, molecular dynamics, and MM-PBSA binding energy calculation of natural alkaloids to get a SARS-CoV-2 Mpro inhibitor
Results and discussion
Virtual screening and molecular docking studies
SARS-CoV-2 Mpro is an enzyme crucial for viral propagation and maturation. Functionally, SARS-CoV-2 Mpro could be a potential drug target to combat COVID-19 [28]. Undoubtedly, nature is a rich source of natural products, such as antiviral agents [29]. Considering the important roles of natural products in the pharma-botanical industry, we performed docking-based virtual screening on natural alkaloids over the SARS-CoV-2 Mpro to identify new inhibitors. Also, drugs currently used for the treatment of COVID-19 patients were used as references to compare the obtained results.
The literature review and structural analysis showed that the active site of SARS-CoV-2 Mpro comprises conserved subunits S1, S2, and S4, and two residues (His 41 and Cys 145) as a catalytic dyad. The S1 subunit serves as an oxyanion hole in the active site and contains residues Phe 140, Ser 144, Cys 145, His 163, and Gln 166. The S2 residues include His 41, Ser 46, Met 49, Asn 51, and Asn 52, while Phe 185, Gln 189, and Gln 192 are located at the S4 position and form a hydrophobic pocket [18, 30, 31]. The docking method was used to identify the inhibitors of SARS-CoV-2 Mpro based on docking score, positions of the ligands in the active site, and their interactions with the key residues of SARS-CoV-2 Mpro. The docking protocol was initially validated via docking the crystal ligand N3 over the SARS-CoV-2 Mpro enzyme. Its poses and interactions were compared with the crystallographic position and interactions available in PDB with a code of 6LU7. As illustrated in Fig. 3a, N3 interacts with the active site residues of SARS-CoV-2 Mpro, such as His 41, Met 49, Phe 140, Cys 145, Met 165, and Gln 189. The calculated RMSD value of N3 for the best-scored conformation was less than 1.98 Å. Also, the docking procedure was validated via the molecular docking study of PF-00835231. PF-00835231 has good interactions with SARS-CoV-2 Mpro active site by establishing hydrogen bonding interactions with residues His 41, Glu166, and Gln 189. Also, it forms hydrophobic interactions with residues Met 49, Lue141, Cys 145, and Met 165 (Fig. 3b). The validated docking procedure was then applied to the selected library of natural alkaloids and FDA-approved antiviral drugs. Each dataset was investigated separately, and the selected compounds were compared and sorted from the lowest to highest docking scores.

Residues involved in the interaction of a N3 and b PF-00835231 with SARS-CoV-2 Mpro according to the docking methodology
As shown in Table 1, all selected drugs exhibited good inhibitory activity against SARS-CoV-2 Mpro except remdesivir. This result is in good agreement with previous studies. Remdesivir, as an adenosine nucleotide analog, is an effective compound for the treatment of COVID-19 by blocking the replication of RNA sequence [32].
As depicted in Fig. 4, nelfinavir and remdesivir occupy the active site by hydrogen bonds and hydrophobic interactions. However, unfavorable bonds (red dotted lines) with His 42 and Asn142 affect its activity stability, and it indicates a repulsion force occurring between remdesivir and SARS-CoV-2 Mpro.

a 2D representation of nelfinavir interaction with SARS-CoV-2 Mpro active site. b Superimpose nelfinavir (slate) and remdesivir (orange) in the active site. c 2D representation of remdesivir interaction in the active site
In order to identify new compounds for counter COVID-19, the docking studies of alkaloids extracted from over 500 medicinal plants and sponges were performed on SARS-CoV-2 Mpro active pocket. Then, compounds were sorted based on docking scores, including the calculated binding energies and their interaction with the key residues of the active pocket of SARS-CoV-2 Mpro. Table 2 shows the molecular docking results for selected natural alkaloids against SARS-CoV-2 Mpro. The calculated binding energies were utilized to rank the selected compounds. Selected compounds (Table 2) could serve as lead compounds for developing the new potential inhibitor candidates against SARS-CoV-2 Mpro. More compounds can be found in Table S1 in the “Supplementary information” section.
Our results indicated that the nine selected natural alkaloids exhibited promising activity against SARS-CoV-2 Mpro (Table 2). It should be noted that compounds have been selected based on docking scores (concerning the lowest binding energy and the best positioning in the active pocket).
Jubanine A, nummularine B, and mucronine D belong to the class of basic macrocyclic compounds known as cyclopeptide alkaloids and are found in the stem bark of Ziziphus jujuba and Zizyphus spina-christi (Fig. 5a) [33]. These compounds are traditionally used to treat or prevent malaria due to their antiplasmodial properties. However, they have not been recommended for COVID-19 patients due to their cytotoxic activity on MRC-5 cells [34]. Virenamide B is a linear thiazole-containing peptide isolated from Diplosoma virens (Fig. 5b). It shows high levels of cytotoxicity by inhibiting microtubule assembly [35]. Therefore, it is not an ideal choice compound. Lasiodine A and asterinin A are linear tetra-peptide, isolated from the leaves of Lasiodiscus marmoratus (Rhamnaceae) and the root of Aster tataicus (Asteraceae), respectively (Fig. 5c). They are identified as new and potent inhibitors of SARS-CoV-2 Mpro (Table 2). However, their biological activity and side effects remain unknown and further studies are required to confirm their applicability for COVID-19 patients. Clionamide, the major metabolite of the sponge cliona celata (Fig. 5d), is used as a mild antibiotic in folk medicine [36]. It deeply penetrates the active pocket of SARS-CoV-2 Mpro and can be a significant resource for developing lead compounds.

Yohimbine, found in the bark of the African tree, Pausinystalia johimbe (Fig. 5e), is a known supplement with alpha-2-adrenergic blocking activity used for weight loss, chest pain, exhaustion, regulation of blood pressure, neuropathic pain in diabetes, depression, and sexual problems [43]. The predicted binding energy and appropriate interactions with the enzyme’s active site suggest that yohimbine is finely docked to the active site of the SARS-CoV-2 Mpro. Yohimbine strongly interacts with SARS-CoV-2 Mpro by forming hydrophobic bonds with His 41, Ser 46, Asn 142, His 163, Glu 166, and Gln 189, and three hydrogen bonds with Leu 141, Ser 144, and Cys 145 (Fig. 6). It should be mentioned that His 41 and Cys 145 play an important role in the catalytic activity of SARS-CoV-2 Mpro [31]. Therefore, it could serve as an appropriate lead compound.

a Superimpose of the yohimbine and ajmalicine with SARS-CoV-2 Mpro active site. Yohimbine and ajmalicine are presented in yellow and green, respectively. b, d 3D and 2D representation of yohimbine interaction; c, e 3D and 2D representation of ajmalicine interaction.
Ajmalicine, naturally found in Catharanthus roseus (Fig. 5f), is structurally related to yohimbine and is used as a drug to treat hypertension and circulatory diseases. It acts as an alpha-adrenergic antagonist and a vasodilator agent [44]. As shown in Fig. 6, ajmalicine deeply penetrated the active pocket of SARS-CoV-2 Mpro and stabilized by forming hydrogen bonds with Gly 143, Ser 144, Cys 145, and His 163. Also, hydrophobic interactions were formed with key residues, including His 41, Lue 141, Asn 142, His 163, Glu 166, and Gln 189, and one pi interaction was formed with Met 49.
Ajmalicine and yohimbine were successfully superimposed in the active pocket of SARS-CoV-2 Mpro (Fig. 6). Docking results showed that yohimbine and ajmalicine occupied the active site by interactions favorably with the S1 and S2 subunits. These alkaloids showed more inhibitory activity against SARS-CoV-2 Mpro than some drugs such as nelfinavir and simeprevir. As expected, the molecular shape of a ligand is tremendously important to reach and fit with the active site. The docking analyses revealed that yohimbine and ajmalicine fit well with the binding site cavity. It shows that the moderate degree of hydrophobicity and forming of hydrogen bonds are crucial to inhibit the activity of SARS-CoV-2 Mpro. Yohimbine and ajmalicine can provide hydrophobic interactions through quinazoline, carbazole, and indole moieties. Also, hydrogen bonds could be formed through hydroxy cyclohexane carboxylate and dihydropyran carboxylate in yohimbine and ajmalicine, respectively.
In summary, based on proper interactions with the active site of SARS-CoV-2 Mpro, low binding energy, few side effects, and their availability in the medicinal market, ajmalicine and yohimbine could be employed as potential therapeutic options for the treatment of COVID-19.
Molecular dynamic simulations
MD simulations have an extensive position in drug discovery since they may explain ligands’ dynamic behavior and stability. Therefore, to specify and compare the stability and behavior of the selected ligands, namely yohimbine and ajmalicine in complex with the SARS-CoV-2 Mpro, MD simulations were employed during 150 ns, and the output was analyzed. Also, the MD simulation became accomplished on SARS-CoV-2 Mpro in the absence of ligands. In order to guarantee the reliability of simulated results, the simulation of all systems was repeated 3 times.
The calculated RMSD (root mean square deviation) values of the backbone atoms of bare protein were compared to that of complex systems to confirm the conformational stability of the protein. As displayed in Fig. 7, yohimbine and ajmalicine complexes attained an equilibration state in less than 20 ns of simulation time. The RMSD plots revealed that the maximum RMSD value, belonging to the bare SARS-CoV-2 Mpro, was approximately 0.3 nm and larger than the complex systems. An interpretation is that the structure of the bare protein is flexible and, therefore, its conformation can change to take a more stable one. It seems that yohimbine has a marked interaction with the active pocket of the protein similar to that of ajmalicine. Because of interactions between the ligands and protein residues, conformational changes in the protein structure were limited, and the RMSD values were decreased in both complexes. Also, the variations in RMSD values were not statistically significant (approximately 0.12 nm in yohimbine and 0.10 nm in ajmalicine), which corroborated the conformational stability of complexes. Furthermore, the repeat simulations yield similar RMSD values, imparting assurance to our observations. Regarding the RMSD profile of ligands (Fig. 8), ajmalicine and yohimbine are structurally stable during all repeat simulations.

The RMSD plots as a function of simulation time. a Repeat simulations of free SARS-CoV-2 Mpro backbone, b repeat simulations of SARS-CoV-2 Mpro backbone in complex with ajmalicine, and c repeat simulations of SARS-CoV-2 Mpro backbone in complex with yohimbine

The RMSD plots as a function of simulation time. a Repeat simulations of ajmalicine, and b repeat simulations of yohimbine
In order to evaluate the variations of protein flexibility and residue fluctuations during the MD simulation period, the RMSF of the backbone residues of both ajmalicine and yohimbine-SARS-CoV-2 Mpro complexes were plotted against each residue number (Fig. 9). A similar RMSF distribution profile was observed in triplicate simulations in both complexes. As depicted in Fig. 9, the residues of the active site, including His 41 and Cys 145, had small fluctuations compared with their adjacent residues. This observation indicated that both ligands interact with the active site residues of SARS-CoV-2 Mpro in relatively the same way. These findings verified the results obtained from the docking analyses and showed that the selected ligands had a significant capability to inhibit SARS-CoV-2 Mpro.

The RMSF plots of ajmalicine and yohimbine-SARS-CoV-2 Mpro complexes from repeat simulations
Calculation of binding free energy
The MM-PBSA method was used to calculate the binding free energy. The results obtained from MM-PBSA calculations are listed in Table 3. Binding free energy calculations showed the high affinity between both ligands and the active pocket of SARS-CoV-2 Mpro. This may be due to the structural similarity of the two compounds. Negative values of van der Waals energies showed that both ligands have appropriate hydrophobic interactions, and they deeply penetrate the hydrophobic part of SARS-CoV-2 Mpro. Also, binding free energy was calculated for binding of remdesivir to SARS-CoV-2 Mpro as a negative control. Remdesivir acts against COVID-19 by blocking the replication of RNA sequence, and therefore, it shows a lower docking score relative to SARS-CoV-2 Mpro inhibitors. Remdesivir exhibits much less binding free energy (− 15.22 kJ.mol−1) than both selected ligands, which confirms the accuracy of the calculations.
Figure 10 shows the calculated binding free energy contribution of the residues involved in interacting with ligands. The results reveal that His 41, Met 165, Gln 189, and Gln 192 are significant participants to the binding energy in both ligands, and Gln 189 was a major contributor for binding to yohimbine with − 9.14 kJ.mol-1. The results obtained from MM-PBSA calculation confirmed that both yohimbine and ajmalicine could occupy the active site of SARS-CoV-2 Mpro.

The total binding energy contribution of amino acids of SARS-CoV-2 Mpro is involved in interaction with ajmalicine (blue) and yohimbine (red)
Conclusions
The rapid, widespread COVID-19 outbreak and lack of proven antiviral drugs have attracted attention to drug discovery’s bioinformatics methods. In this study, we conducted systematic docking-based virtual screening on natural alkaloids to search for potential inhibitors of SARS-CoV-2 Mpro. Subsequently, repeat molecular dynamic simulations followed by binding free energy calculations were applied to determine the stability and inhibitory potential of selected ligands in the binding pocket of SARS-CoV-2 Mpro in 150 ns. Based on the binding affinity and interactions with active site residues, ajmalicine and yohimbine were identified as the most potent inhibitors among several natural alkaloids. The results of the MD simulations, such as analyzing the stability and binding free energies, confirmed that selected ligands with high docking scores retain their interaction and stability over time. Therefore, they could be regarded as lead compounds for developing inhibitors of SARS-CoV-2 Mpro. We believe that these indole alkaloids will enrich the armamentarium of therapeutic agents to fight COVID-19. Further analyses are warranted to elucidate the mechanisms of action of these agents.
Data availability
All data generated or analyzed during this investigation are included in this published article.
Code availability
There are no codes used in the present investigation.
References
Baden LR, Rubin EJ (2020) Covid-19 — the search for effective therapy. N Engl J Med 382(19):1851–1852. https://doi.org/10.1056/NEJMe2005477
Mousavizadeh L, Ghasemi S (2020) Genotype and phenotype of COVID-19: their roles in pathogenesis. J Microbiol Immunol Infect 54(2):159–163. https://doi.org/10.1016/j.jmii.2020.03.022
Sharifkashani S, Bafrani MA, Khaboushan AS, Pirzadeh M, Kheirandish A, Yavarpour Bali H, Hessami A, Saghazadeh A, Rezaei N (2020) Angiotensin-converting enzyme 2 (ACE2) receptor and SARS-CoV-2: potential therapeutic targeting. Eur J Pharmacol 884:173455. https://doi.org/10.1016/j.ejphar.2020.173455
V’kovski P, Kratzel A, Steiner S, Stalder H, Thiel V (2020) Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol 19:155–170. https://doi.org/10.1038/s41579-020-00468-6
Gupta SP (2017) Viral proteases and their inhibitors. 1st Edition, Academic Press.
Keretsu S, Bhujbal SP, Cho SJ (2020) Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci Rep 10(1):17716. https://doi.org/10.1038/s41598-020-74468-0
Cui Q, Du R, Liu M, Rong L (2020) Lignans and their derivatives from plants as antivirals. Molecules 25(1):183. https://doi.org/10.3390/molecules25010183
Ghosh R, Chakraborty A, Biswas A, Chowdhuri S (2021) Identification of alkaloids from Justicia adhatoda as potent SARS CoV-2 main protease inhibitors: an in silico perspective. J Mol Struct 1229:129489. https://doi.org/10.1016/j.molstruc.2020.129489
Qing Z-X, Yang P, Tang Q, Cheng P, Liu X-B, Zheng Y-j, Liu Y-S, Zeng J-G (2017) Isoquinoline alkaloids and their antiviral, antibacterial, and antifungal activities and structure-activity relationship. Curr Org Chem 21(18):1920–1934. https://doi.org/10.2174/1385272821666170207114214
Xu W, Zhang M, Liu H, Wei K, He M, Li X, Hu D, Yang S, Zheng Y (2019) Antiviral activity of aconite alkaloids from Aconitum carmichaelii Debx. Nat Prod Res 33(10):1486–1490. https://doi.org/10.1080/14786419.2017.1416385
Bahrami H, Salehabadi H, Nazari Z, Amanlou M (2019) Combined virtual screening, DFT calculations and molecular dynamics simulations to discovery of potent MMP-9 inhibitors. Lett Drug Des Discov 16(8):892–903. https://doi.org/10.2174/1570180815666181008095950
Wang J (2020) Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. J Chem Inf Model 60(6):3277–3286. https://doi.org/10.1021/acs.jcim.0c00179
Sattari A, Ramazani A, Aghahosseini H (2021) Repositioning therapeutics for COVID-19: virtual screening of the potent synthetic and natural compounds as SARS-CoV-2 3CLpro inhibitors. J Iran Chem Soc 18:2807–2827. https://doi.org/10.1007/s13738-021-02235-7
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30(16):2785–2791. https://doi.org/10.1002/jcc.21256
Buckingham J, Baggaley KH, Roberts AD, Szabo LF (2010) Dictionary of alkaloids with CD-ROM. 2nd Edition, CRC Press.
Kapoor R, Sharma B, Kanwar SS (2017) Antiviral phytochemicals: an overview Biochem Physiol 6:220. https://doi.org/10.4172/2168-9652.1000220
Bastida J, Viladomat F, Codina C (1997) Narcissus alkaloids. Studies in Natural Products Chemistry. Part F, Elsevier 20:323–405. https://doi.org/10.1016/S1572-5995(97)80034-8
Hosseini FS, Amanlou M (2020) Anti-HCV and anti-malaria agent, potential candidates to repurpose for coronavirus infection: virtual screening, molecular docking, and molecular dynamics simulation study. Life sci 258:118205. https://doi.org/10.1016/j.lfs.2020.118205
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open Babel: an open chemical toolbox. J Cheminformatics 3(1):33. https://doi.org/10.1186/1758-2946-3-33
de Vries M, Mohamed AS, Prescott RA, Valero-Jimenez AM, Desvignes L, O’Connor R, Steppan C, Devlin JC, Ivanova E, Herrera A, Schinlever A, Loose P, Ruggles K, Koralov SB, Anderson AS, Binder J, Dittmann M (2021) A comparative analysis of SARS-CoV-2 antivirals characterizes 3CLpro inhibitor PF-00835231 as a potential new treatment for COVID-19. J Virol 95(10):e01819-e1820. https://doi.org/10.1128/JVI.01819-20
Discovery Studio Modeling Environment, Release 2017. BIOVIA, Dassault Systèmes, San Diego.
Makarewicz T, Kaźmierkiewicz R (2013) Molecular dynamics simulation by GROMACS using GUI plugin for PyMOL. J Chem Inf Model 53(5):1229–1234. https://doi.org/10.1021/ci400071x
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2:19–25. https://doi.org/10.1016/j.softx.2015.06.001
Sousa da Silva AW, Vranken WF (2012) ACPYPE - AnteChamber PYthon Parser interfacE. BMC Res Notes 5(1):367. https://doi.org/10.1186/1756-0500-5-367
Søndergaard CR, Olsson MHM, Rostkowski M, Jensen JH (2011) Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J Chem Theory comput 7(7):2284–2295. https://doi.org/10.1021/ct200133y
Kumari R, Kumar R, Consortium OSDD, Lynn A (2014) g_mmpbsa—a GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54(7):1951–1962. https://doi.org/10.1021/ci500020m
Hassanzadeh M, Bagherzadeh K, Amanlou M (2016) A comparative study based on docking and molecular dynamics simulations over HDAC-tubulin dual inhibitors. J Mol Graph Model 70:170–180. https://doi.org/10.1016/j.jmgm.2016.10.007
Paul D, Basu D, Ghosh Dastidar S (2021) Multi-conformation representation of Mpro identifies promising candidates for drug repurposing against COVID-19. J Mol Model 27:128. https://doi.org/10.1007/s00894-021-04732-1
Martinez JP, Sasse F, Brönstrup M, Diez J, Meyerhans A (2015) Antiviral drug discovery: broad-spectrum drugs from nature. Nat Prod Rep 32(1):29–48. https://doi.org/10.1039/C4NP00085D
Bharadwaj S, Lee KE, Dwivedi VD, Kang SG (2020) Computational insights into tetracyclines as inhibitors against SARS-CoV-2 Mpro via combinatorial molecular simulation calculations. Life sci 257:118080. https://doi.org/10.1016/j.lfs.2020.118080
Günther S, Reinke PY, Fernández-García Y et al (2021) X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 372(6542):642–646. https://doi.org/10.1126/science.abf7945
Agostini ML, Andres EL, Sims AC, Graham RL, Sheahan TP, Lu X, Smith EC, Case JB, Feng JY, Jordan R, Ray AS, Cihlar T, Siegel D, Mackman RL, Clarke MO, Baric RS, Denison MR (2018) Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. MBio 9(2):e00221–e00218. https://doi.org/10.1128/mBio.00221-18
Abdel-galil FM, El-Jissry MA (1991) Cyclopeptide alkaloids from Zizyphus spina-christi. Phytochemistry 30(4):1348–1349. https://doi.org/10.1016/S0031-9422(00)95238-5
Tuenter E, Segers K, Kang KB, Viaene J, Sung SH, Cos P, Maes L, Heyden YV, Pieters L (2017) Antiplasmodial activity, cytotoxicity and structure-activity relationship study of cyclopeptide alkaloids. Molecules 22(2):224. https://doi.org/10.3390/molecules22020224
Tran TD, Pham NB, Fechner GA, Hooper JNA, Quinn RJ (2014) Potent cytotoxic peptides from the Australian marine sponge Pipestela candelabra. Mar Drugs 12(6):3399–3415. https://doi.org/10.3390/md12063399
Kumar D, Arya V, Kaur R, Bhat ZA, Gupta VK, Kumar V (2012) A review of immunomodulators in the Indian traditional health care system. J Microbiol Immunol Infect 45(3):165–184. https://doi.org/10.1016/j.jmii.2011.09.030
T. Mostafavi, Properties of jujube for weight loss, accessed 21 October 2021, <http://av5.ir/6v3>.
S. Leyte, Diplosoma virens, (Family Didemnidae), accessed 21 October 2021, <http://www.wetwebmedia.com/SWPOTD595.htm>.
Aster tataricus, Wikimedia Commons, the free media repository, accessed 21 October 2021, <https://commons.wikimedia.org/wiki/File:Aster_tataricus1.jpg>.
B. E. Picton, C. C. Morrow, R. W. B. van Soest, Cliona celata, Sponges of Britain and Ireland, accessed 21 October 2021, <http://www.habitas.org.uk/marinelife/sponge_guide/sponges.asp?item=C3020>.
Pausinystalia johimbe.jpg, PsychonautWiki, accessed 21 October 2021, <https://psychonautwiki.org/wiki/File:Pausinystalia_johimbe.jpg>.
Catharanthus roseus24 08 2012 (1).JPG, Wikimedia Commons, the free media repository, accessed 21 October 2021, <https://en.wikipedia.org/wiki/Catharanthus_roseus#/media/File:Catharanthus_roseus24_08_2012_(1).JPG>.
Cohen PA, Wang Y-H, Maller G, DeSouza R, Khan IA (2016) Pharmaceutical quantities of yohimbine found in dietary supplements in the USA. Drug Test Anal 8:357–369. https://doi.org/10.1002/dta.1849
Mendonça Freitas MS, Gama MC, Monnerat PH, De Carvalho AJC, Lima TC, Vieira IJC (2016) Induced nutrient deficiencies in Catharanthus roseus impact ajmalicine bioproduction. J Plant Nutr 39(6):835–841. https://doi.org/10.1080/01904167.2015.1047524
Funding
This work has been supported financially by the Research Deputy of Zanjan University of Medical Sciences, Zanjan, Iran (Grant No. A-11–1510-1).
Author information
Authors and Affiliations
Contributions
M.M. and B.F. generated the idea and collected the initial library of alkaloids. H.B. provided critical input and suggestions in writing the manuscript’s discussion part and prepared figures. H.S. conducted the virtual screening, molecular docking, and molecular dynamic simulation experiments and wrote the initial draft of the manuscript. M.A. and F.S.H. critically reviewed the idea generation and providing computational resources. All the authors read and approved the final draft of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

{kind=link}
{kind=link}
.JPG){kind=link}
Cite this article
Mohseni, M., Bahrami, H., Farajmand, B. et al. Indole alkaloids as potential candidates against COVID-19: an in silico study. J Mol Model 28, 144 (2022). https://doi.org/10.1007/s00894-022-05137-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-022-05137-4