
Abstract
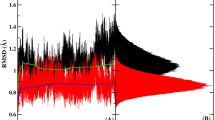
Thrombin is a Na\(^+\)-activated serine protease existing in two forms targeted to procoagulant and anticoagulant activities, respectively. There is one Na\(^+\)-binding site that has been the focus of the study of the thrombin. However, molecular dynamics (MD) simulations suggest that there might be actually two Na\(^+\)-binding sites in thrombin and that Na\(^+\) ions can even bind to two sites simultaneously. In this study, we performed 12 independent 2-µs all-atom MD simulations for the wild-type (WT) thrombin and we studied the effects of the different Na\(^+\) binding modes on thrombin. From the root-mean-square fluctuations (RMSF) for the \(\alpha\)-carbons, we see that the atomic fluctuations mainly change in the 60s, 170s, and 220s loops, and the connection (residue 167 to 170). The correlation matrices for different binding modes suggest regions that may play an important role in thrombin’s allosteric response and provide us a possible allosteric pathway for the sodium binding. Amorim-Hennig (AH) clustering tells us how the structure of the regions of interest changes on sodium binding. Principal component analysis (PCA) shows us how the different regions of thrombin change conformation together with sodium binding. Solvent-accessible surface area (SASA) exposes the conformational change in exosite I and catalytic triad. Finally, we argue that the double binding mode might be an inactive mode and that the kinetic scheme for the Na\(^+\) binding to thrombin might be a multiple-step mechanism rather than a 2-step mechanism.










Similar content being viewed by others

Data availability
Data available by request.
Code availability
Code available by request.
References
Abdi H, Williams LJ (2010) Principal component analysis. Wiley Interdiscip Rev Comput Stat 2(4), 433–459
Bah A, Garvey LC, Ge J, Di Cera E (2006) Rapid kinetics of na+ binding to thrombin. Journal of Biological Chemistry 281(52), 40049–40056
Bah A, Chen Z, Bush-Pelc LA, Mathews FS, Di Cera E (2007) Crystal structures of murine thrombin in complex with the extracellular fragments of murine protease-activated receptors par3 and par4. Proceedings of the National Academy of Sciences 104(28), 11603–11608
Berendsen HJ, Postma Jv, van Gunsteren WF, DiNola A, Haak JR (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81(8), 3684–3690
Bode W, Turk D, Karshikov A (1992) The refined 1.9-å x-ray crystal structure of d-phe-pro-arg chloromethylketone-inhibited human \(\alpha\)-thrombin: Structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure-function relationships. Protein science 1(4):426–471
Bro R, Smilde AK (2014) Principal component analysis. Analytical methods 6(9):2812–2831
Carrell CJ, Bush LA, Mathews FS, Di Cera E (2006) High resolution crystal structures of free thrombin in the presence of k+ reveal the molecular basis of monovalent cation selectivity and an inactive slow form. Biophysical chemistry 121(3):177–184
Carter WJ, Myles T, Gibbs CS, Leung LL, Huntington JA (2004) Crystal structure of anticoagulant thrombin variant e217k provides insights into thrombin allostery. Journal of Biological Chemistry 279(25), 26387–26394
Darden T, York D, Pedersen L (1993) Particle mesh ewald: An N Log (n) method for ewald sums in large systems. J Chem Phys 98(12), 10089–10092
Davie EW, Kulman JD (2006) An overview of the structure and function of thrombin. In: Seminars in thrombosis and hemostasis, Copyright 2006 by Thieme Medical Publishers, Inc., 333 Seventh Avenue, New ..., vol 32, pp 003–015
De Amorim RC, Hennig C (2015) Recovering the number of clusters in data sets with noise features using feature rescaling factors. Inf Sci (N Y) 324:126–145
Di Cera E (2008) Thrombin. Mol Aspects Med 29(4), 203–254
Di Cera E, Guinto ER, Vindigni A, Dang QD, Ayala YM, Wuyi M, Tulinsky A (1995) The na+ binding site of thrombin. Journal of Biological Chemistry 270(38), 22089–22092
Feenstra KA, Hess B, Berendsen HJ (1999) Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J Comput Chem 20(8), 786–798
Fuglestad BA (2013) Exploring the Dynamics of Thrombin by NMR. University of California, San Diego
Gandhi PS, Chen Z, Di Cera E (2010) Crystal structure of thrombin bound to the uncleaved extracellular fragment of par1. Journal of Biological Chemistry 285(20), 15393–15398
Godwin R, Gmeiner W, Salsbury Jr FR (2016) Importance of long-time simulations for rare event sampling in zinc finger proteins. J Biomol Struct Dyn 34(1), 125–134
Godwin RC, Melvin R, Salsbury FR (2015) Molecular dynamics simulations and computer-aided drug discovery. In: Computer-aided drug discovery, Springer, pp 1–30
Godwin RC, Melvin RL, Gmeiner WH, Salsbury Jr FR (2017) Binding site configurations probe the structure and dynamics of the zinc finger of nemo (nf-kb essential modulator). Biochemistry 56(4), 623–633
Handley LD, Treuheit NA, Venkatesh VJ, Komives EA (2015) Thrombomodulin binding selects the catalytically active form of thrombin. Biochemistry 54(43), 6650–6658
Handley LD, Fuglestad B, Stearns K, Tonelli M, Fenwick RB, Markwick PR, Komives EA (2017) Nmr reveals a dynamic allosteric pathway in thrombin. Scientific reports 7(1):1–9
Harvey M, De Fabritiis G (2009) An implementation of the smooth particle mesh ewald method on gpu hardware. J Chem Theory Comput 5(9), 2371–2377
Harvey MJ, Giupponi G, Fabritiis GD (2009) Acemd: Accelerating biomolecular dynamics in the microsecond time scale. J Chem Theory Comput 5(6), 1632–1639
Humphrey W, Dalke A, Schulten K (1996) Vmd: Visual molecular dynamics. J Mol Graphics 14(1), 33–38
Huntington JA, Esmon CT (2003) The molecular basis of thrombin allostery revealed by a 1.8 å structure of the “slow” form. Structure 11(4):469–479
Ichiye T, Karplus M (1991) Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins: Structure, Function, and Bioinformatics 11(3):205–217
Johnson DJ, Adams TE, Li W, Huntington JA (2005) Crystal structure of wild-type human thrombin in the na+-free state. Biochemical Journal 392(1), 21–28
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79(2), 926–935
Kurisaki I, Takayanagi M, Nagaoka M (2015) Toward understanding allosteric activation of thrombin: A conjecture for important roles of unbound na+ molecules around thrombin. The Journal of Physical Chemistry B 119(9), 3635–3642
Lai MT, Di Cera E, Shafer JA (1997) Kinetic pathway for the slow to fast transition of thrombin: Evidence of linked ligand binding at structurally distinct domains. Journal of Biological Chemistry 272(48), 30275–30282
Lemons DS, Gythiel A (1997) Paul langevin’s 1908 paper “on the theory of brownian motion”[“sur la théorie du mouvement brownien,” cr acad. sci.(paris) 146, 530–533 (1908)]. Am J Phys 65(11):1079–1081
MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102(18), 3586–3616
Mackerell Jr AD, Feig M, Brooks III CL (2004) Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem 25(11), 1400–1415
Maragoudakis ME, Tsopanoglou NE (2010) Thrombin: physiology and disease, vol 19. Springer Science & Business Media
Melvin R, Salsbury F (2016) Hdbscan and amorim-hennig for md. Figshare 10:m9
Melvin RL, Salsbury Jr FR (2016) Visualizing ensembles in structural biology. J Mol Graphics Modell 67:44–53
Melvin RL, Godwin RC, Xiao J, Thompson WG, Berenhaut KS, Salsbury Jr FR (2016) Uncovering large-scale conformational change in molecular dynamics without prior knowledge. J Chem Theory Comput 12(12), 6130–6146
Melvin RL, Thompson WG, Godwin RC, Gmeiner WH, Salsbury Jr FR (2017) Muts\(\alpha\)’s multi-domain allosteric response to three dna damage types revealed by machine learning. Front Phys(Lausanne) 5:10
Nayal M, Di Cera E (1996) Valence screening of water in protein crystals reveals potential na+ binding sites
Papaconstantinou ME, Carrell CJ, Pineda AO, Bobofchak KM, Mathews FS, Flordellis CS, Maragoudakis ME, Tsopanoglou NE, Di Cera E (2005) Thrombin functions through its rgd sequence in a non-canonical conformation. Journal of Biological Chemistry 280(33), 29393–29396
Pineda AO, Carrell CJ, Bush LA, Prasad S, Caccia S, Chen ZW, Mathews FS, Di Cera E (2004a) Molecular dissection of na+ binding to thrombin. Journal of Biological Chemistry 279(30):31842–31853
Pineda AO, Chen ZW, Caccia S, Cantwell AM, Savvides SN, Waksman G, Mathews FS, Di Cera E (2004b) The anticoagulant thrombin mutant w215a/e217a has a collapsed primary specificity pocket. Journal of Biological Chemistry 279(38):39824–39828
Pineda AO, Chen ZW, Bah A, Garvey LC, Mathews FS, Di Cera E (2006) Crystal structure of thrombin in a self-inhibited conformation. Journal of Biological Chemistry 281(43), 32922–32928
Prasad S, Cantwell AM, Bush LA, Shih P, Xu H, Di Cera E (2004) Residue asp-189 controls both substrate binding and the monovalent cation specificity of thrombin. Journal of Biological Chemistry 279(11), 10103–10108
Rousseeuw PJ (1987) Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J Comput Appl Math 20:53–65, 10.1016/0377-0427(87)90125-7, URL http://www.sciencedirect.com/science/article/pii/0377042787901257
Russo Krauss I, Merlino A, Randazzo A, Novellino E, Mazzarella L, Sica F (2012) High-resolution structures of two complexes between thrombin and thrombin-binding aptamer shed light on the role of cations in the aptamer inhibitory activity. Nucleic Acids Res 40(16), 8119–8128
Šali A, Blundell TL (1993) Comparative Protein Modelling by Satisfaction of Spatial Restraints. Journal of Molecular Biology 234(3):779–815, 10.1006/jmbi.1993.1626, URL http://linkinghub.elsevier.com/retrieve/pii/S0022283683716268
Scherer MK, Trendelkamp-Schroer B, Paul F, Pérez-Hernández G, Hoffmann M, Plattner N, Wehmeyer C, Prinz JH, Noé F (2015) Pyemma 2: A software package for estimation, validation, and analysis of markov models. J Chem Theory Comput 11(11), 5525–5542
Van Gunsteren W, Berendsen HJ (1977) Algorithms for macromolecular dynamics and constraint dynamics. Mol Phys 34(5), 1311–1327
Vogt AD, Bah A, Di Cera E (2010) Evidence of the E*- E equilibrium from rapid kinetics of na+ binding to activated protein c and factor xa. The Journal of Physical Chemistry B 114(49), 16125–16130
Workman Jr EF, Lundblad RL (1978) The effect of monovalent cations on the catalytic activity of thrombin. Archives of biochemistry and biophysics 185(2):544–548
Wu D, Xiao J, Salsbury Jr FR (2021) Light chain mutation effects on the dynamics of thrombin. Journal of Chemical Information and Modeling 61(2), 950–965
Xiao J, Salsbury FR (2017) Molecular dynamics simulations of aptamer-binding reveal generalized allostery in thrombin. J Biomol Struct Dyn 35(15), 3354–3369
Xiao J, Salsbury FR (2019) Na+-binding modes involved in thrombin’s allosteric response as revealed by molecular dynamics simulations, correlation networks and markov modeling. Phys Chem Chem Phys 21(8), 4320–4330
Xiao J, Melvin RL, Salsbury FR (2017) Mechanistic insights into thrombin’s switch between “slow” and “fast” forms. Phys Chem Chem Phys 19(36), 24522–24533
Xiao J, Melvin RL, Salsbury Jr FR (2019) Probing light chain mutation effects on thrombin via molecular dynamics simulations and machine learning. J Biomol Struct Dyn 37(4), 982–999
Zhang E, Tulinsky A (1997) The molecular environment of the na+ binding site of thrombin. Biophysical chemistry 63(2–3):185–200
Acknowledgements
The authors wish to acknowledge the support of the Wake Forest Baptist Comprehensive Cancer Center Crystallography & Computational Biosciences Shared Resource, supported by the National Cancer Institute’s Cancer Center Support Grant award number P30CA012197. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute. Some computations were performed on the Wake Forest University DEAC Cluster, a centrally managed resource with support provided in part by the University. FRS would also like to thank the Scott Family for the Scott Family Fellowship.
Funding
Partial support by NIH P30CA012197, Scott Family Fellowship.
Author information
Authors and Affiliations
Contributions
DW performed the calculations, FRS oversaw the project, and DW and FRS conceived of the project and developed the analyses.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary information
Supplementary information
The following format should be used: Supporting Information Available: [Sodium ion coordinated by TYR184 and five water molecules; RMSF with standard errors for unbound, double binding, outer binding, and inner binding, respectively; IMWKRescaled clustering visualization for the \(\gamma\) loop; IMWKRescaled clustering visualization for the catalytic triad; Time series plots of the alpha carbons’ root-mean-square deviation (RMSD) referring to the initial structure of the thrombin; The probability of the closest mean distance between Na\(^+\) ions and the 220s loop; Sodium ion coordinated by O\(_1\)-O\(_5\); Correlation coefficients for Figure 4e–g.]
Rights and permissions
About this article

Cite this article
Wu, D., Salsbury, F.R. Simulations suggest double sodium binding induces unexpected conformational changes in thrombin. J Mol Model 28, 120 (2022). https://doi.org/10.1007/s00894-022-05076-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-022-05076-0