
Abstract
The association of Cu–X (X = –H, –Cl, and –F) with H2CCHCHmYn and HCCCHmYn (Y = –Cl, –F, –OH, –CH3) has been studied at a high level of theory. The density functional theory (DFT) at B3LYP/6-311G(d,p)//B3LYP/6-311 + G(3df,2p) level has been chosen to calculate the structure and the relative stability of 24 different complexes. The interaction of Cu–F with the derivatives of ethylene and acetylene was found very strong, with interaction energies close to those of conventional covalent bonds. In all complexes, the most stable structure was found when Cu–X is positioned on the unsaturated CC bond, forming a three-membered ring that leads to longer CC bond distances. Both ethylene and acetylene complexes show similar trends of interaction energies with respect to the same moiety. All electronic indexes analyzed by means of the QTAIM, ELF, and NBO formalisms indicate that the strength of the interaction should increase with the number of withdrawing substituents in both series of compounds.

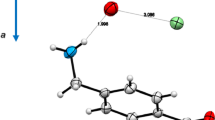
The p-Interaction of ethylene and acetylene derivative with fluoride copper. The ELF graphs and its 2D projection show the disynaptic basins of the electrostatic binding








Similar content being viewed by others

References
Du X, Fan R, Wang X, Qiang L, Wang P, Gao S, Zhang H, Yang Y, Wang Y (2015) Combined effect of hydrogen bonding and π···π stacking interactions in the assembly of indium(III) metal-organic materials: structure-directing and aggregation-induced emission behavior. Crystal Growth and Design 15(5):2402–2412
Moncho S, Brothers EN, Hall MB (2015) Addition of ethylene to a π-conjugated two-dimensional nickel-based organometallic framework with implications for olefin separation. J. Mol. Mod. 21(5)
Sakamoto R, Wu KH, Matsuoka R, Maeda H, Nishihara H (2015) π-Conjugated bis(terpyridine)metal complex molecular wires. Chem. Soc. Rev. 44(21):7698–7714
Yamabe K, Nomura N, Goto H (2019) Biological quorum sensing molecule-metal complex produces π-conjugated polymer. Inter. J. Poly. Mat. Poly. Biomat. 68(13):805–809
Barrientos L, Miranda-Rojas S, Mendizabal F (2019) Noncovalent interactions in inorganic supramolecular chemistry based in heavy metals. Quantum chemistry point of view. Int. J. Quant. Chem. 119(2)
Oliveira V, Cremer D (2017) Transition from metal-ligand bonding to halogen bonding involving a metal as halogen acceptor a study of Cu, Ag, Au, Pt, and Hg complexes. Chem. Phys. Lett. 681:56–63
Lamsabhi AM, Alcamí M, Mó O, Yáñez M (2003) Gas-phase reactivity of uracil, 2-thiouracil, 4-thiouracil, and 2,4-dithiouracil towards the Cu+ cation: a DFT study. ChemPhysChem 4(9):1011–1016
Lamsabhi AM, Alcamí M, Mó O, Yáñez M, Tortajada J (2004) Association of Cu2+ with uracil and its thio derivatives: a theoretical study. ChemPhysChem 5(12):1871–1878
Lamsabhi AM, Alcamí M, Mó O, Yáñez M, Tortajada J (2006) Gas-phase deprotonation of uracil-Cu2+ and thiouracil-Cu2+ complexes. J. Phys. Chem. A 110(5):1943–1950
Kaur K, Sharma A, Capalash N, Sharma P (2019) Multicopper oxidases: biocatalysts in microbial pathogenesis and stress management. Microbiol. Res. 222:1–13
Abed F, Rayah H, Bouamrane R, Al-Taiar AH (2015) Spectroscopic studies of charge transfer complexes of some amino acids with copper (II). Oriental J. Chem. 31(1):493–497
De Gregorio G, Biasotto F, Hecel A, Luczkowski M, Kozlowski H, Valensin D (2019) Structural analysis of copper(I) interaction with amyloid β peptide. J. Inorg. Biochem. 195:31–38
Li XW, Zhao XH, Li YT, Wu ZY (2019) Synthesis and crystal structure of bicopper(II) complexes: the influence of bridging ligands on DNA/BSA binding behaviors and in vitro antitumor activity. Inorg. Chim. Acta 488:219–228
Arslancan S, Lamsabhi AM, Mó O, Yáñez M (2018) Complexes between cyclopentene and cyclopentyne derivatives with HCu and FCu: the importance of cyclization effects. Int. J. Quantum Chem. 118(9):e25489
Sánchez-Sanz G, Alkorta I, Elguero J, Yáñez M, Mó O (2012) Strong interactions between copper halides and unsaturated systems: new metallocycles? Or the importance of deformation. Phys. Chem. Chem. Phys. 14(32):11468
Becke AD (1993) Density-functional thermochesmistry. 3. The role of exact exchange. J. Chem. Phys. 98(7):5648–5652
Lee CT, Yang WT, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 37(2):785–789
Inada Y, Orita H (2007) Efficiency of numerical basis sets for predicting the binding energies of hydrogen bonded complexes: evidence of small basis set superposition error compared to Gaussian basis sets. J. Comput. Chem., vol 29. John Wiley & Sons, Ltd.
Kobko N, Dannenberg JJ (2001) Effect of basis set superposition error (BSSE) upon ab initio calculations of organic transition states. J. Phys. Chem. A 105(10):1944–1950
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Frisch H, Trucks MJ, Schlegel GW, Scuseria HB, Robb GE, Cheeseman MA, Scalmani JR, Barone G, Mennucci V, Petersson B, Nakatsuji GA, Caricato H, Li M, Hratchian X, Izmaylov HP, Bloino AF, Zheng J, Sonnenberg G, Hada JL, Ehara M, Toyota M, Fukuda K, Hasegawa R, Ishida J, Nakajima M, Honda T, Kitao Y, Nakai O, Vreven H, Montgomery TJ, Peralta JA, Ogliaro JE, Bearpark F, Heyd M, Brothers JJ, Kudin E, Staroverov KN, Kobayashi VN, Normand R, Raghavachari J, Rendell K, Burant A, Iyengar JC, Tomasi SS, Cossi J, Rega M, Millam N, Klene JM, Knox M, Cross JE, Bakken JB, Adamo V, Jaramillo C, Gomperts J, Stratmann R, Yazyev RE, Austin O, Cammi AJ, Pomelli R, Ochterski C, Martin JW, Morokuma RL, Zakrzewski K, Voth VG, Salvador GA, Dannenberg P, Dapprich JJ, Daniels S, Farkas AD, Foresman Ö, Ortiz JB, Cioslowski JV, Fox J (2017) Gaussian 09. Revision E01 edn. Gaussian Inc,, Wallingford, CT
Bader RFW (1990) Atoms in molecules. Clarendon Press, Oxford
Matta CF, Boyd RJ (eds) (2007) The quantum theory of atoms in molecules: from solid state to DNA and drug design. Wiley-VCH Verlag, Weinheim
Becke AD, Edgecombe KE (1990) A simple measure of electron localization n atomic and molecular-systems. J. Chem. Phys. 92:5397–5403
Weinhold F, R Landis C (2005) Valency and bonding
Keith TA (2018) AIMAll http://aim.tkgristmill.com 17.11.14 edn., Overland Park
Glendening ED, Badenhoop JK, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Landis CR, Weinhold F (2013) NBO http://nbo6.chem.wisc.edu/. 6.0 edn., Theoretical chemistry institute, University of Wisconsin, Madison
Silvi B (2010) TopMod http://www.lct.jussieu.fr/pagesperso/silvi/topmod.html. GNU edn. The TopMod
Ziegler T, Rauk A (1977). Theor. Chim. Acta 46:1–10
Bickelhaupt FM, Baerends EJ (2007) Kohn-Sham density functional theory: predicting and understanding chemistry. In: Lipkowitz KB, Boyd DB (eds) Reviews in computational chemistry. John Wiley & Sons, Inc., pp 1–86
Kitaura K, Morokuma K . Int. J. Quant. Chem. 10:325 (1976)
A. Vu Theoretical chemistry in Amsterdam density functional (ADF) program, http://www.scm.com.(2016)
Acknowledgments
This work has been partially supported by the FONDECYT REGULAR 1170837 (BH) DGI Projects no. CTQ2015-63997-C2. A generous allocation of computing time at the Centro de Computación Científica of the UAM is also acknowledged (AML).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 25.4 kb)
Rights and permissions
About this article

Cite this article
Arslancan, S., Herrera, B. & Lamsabhi, A.M. On the nature of the interaction of copper hydride and halide with substituted ethylene and acetylene. J Mol Model 26, 61 (2020). https://doi.org/10.1007/s00894-020-4320-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-020-4320-0