
Abstract
The gas phase mechanism of the peptide bond formation between two double amino acid (DAA) molecules described by the (NH2)2C(COOH)2 formula is investigated in the presence of a water molecule. Formations of trans and cis DAA–DAA dipeptide products along both concerted and stepwise mechanisms have been studied at the CCSD(T)/aug-cc-pVDZ//MP2/aug-cc-pVDZ level. The results indicate that the activation energy barriers estimated for the water-assisted mechanisms are significantly reduced in comparison to the corresponding uncatalyzed reactions. The trans DAA–DAA isomer is expected to dominate in the final product due to its larger stability compared to the cis DAA–DAA product.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Condensation of amino acids is a crucial reaction in protein chemistry as it represents a key reaction for all life processes. An essential structural element of all proteins is a peptide bond (C–N), which is formed because of the conjugation between the α-amino group of one amino acid and the α-carboxylic group of another amino acid. The detailed knowledge of the peptide bond formation mechanism is vital for understanding of various biological processes, and it is a subject of intensive investigations as such. The uncatalyzed reaction of peptide bond formation has been studied by Oie et al. [1] and Jensen et al. [2], who predicted two possible mechanisms, namely concerted and stepwise, and summarized that these two routes may compete since both require similar activation energies (relative energies of the highest transition state structure with respect to the reactants are about 40–47 kcal mol–1). More recently, similar results have been obtained by Redondo et al. [3] and Dornshuld et al. [4], who performed the investigation of glycine condensation in the absence of any catalysts.
Typical high-yielding reactions of peptide bond formation require the presence of an activated amino acid precursor [5]. On the other hand, the synthesis of peptide bond by direct condensation of unactivated amino acids is a very slow process, and the presence of a suitable catalyst is needed in order to overcome kinetic limitation [6]. The catalytic role in the formation of polypeptides can be played by minerals [7], mineral and metal oxide surfaces [8,9,10,11], metal dications (e.g., Mg2+, Ca2+, Cu2+ or Ni2+) [12,13,14,15,16], and protic solvents (such as ammonia [17] and water [18]). Theoretical calculations have also been proven useful in this field as providing an insight into such mechanisms at the molecular level. For example, Oie et al. [19] employed computational methods to study the amine catalyzed peptide bond formation between formic acid and an ammonia molecule and demonstrated that the activation barrier was reduced by about 10 kcal mol–1 with respect to the uncatalyzed process. Even more promising results were obtained by Rimola et al., who theoretically investigated the effect of the simultaneous presence of Lewis and Brønsted catalysts (AlF3 and HF, respectively) on the same reaction. Namely, they found that the synergy between these catalysts reduces the activation energy barrier from 56.0 kcal mol–1 (uncatalyzed reaction) to 14.4 kcal mol–1 [20]. Since the water molecules are generated during the amino acids condensation, earlier computational studies were also focused on the catalytic role that H2O plays in the Ala–Ala peptide bond formation [18] and on the generation of cyclic dipeptide from various amino acids [21]. In both these cases, the barrier heights were found to be systematically reduced owing to the assistance of water molecules. The same effect was observed for very similar process, namely the ammonia addition to formaldehyde assisted by water, which has been investigated by Williams [22].
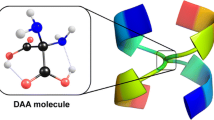
In this contribution, we explore the gas phase mechanism of the peptide bond formation between two double amino acid (DAA) molecules in the presence of a water molecule. The double amino acid (described by the (NH2)2C(COOH)2 formula, see Fig. 1) has been recently proposed by our group as a novel amino acid enabling peptide interpenetrating structures [23]. Its stability with respect to both unimolecular deamination and decarboxylation reactions has already been addressed in order to show that spontaneous detachment of either ammonia or the carbon dioxide molecule in gas and aqueous phases is not operative [24, 25]. Moreover, we demonstrated that the DAA system acts similarly to various natural amino acids when the noncatalyzed peptide bond formation process between two DAA molecules or the cyclization involving two DAAs in gas phase are considered [26, 27].

Equilibrium structure of the most stable canonical isomer of double amino acid (DAA) molecule in the gas phase
Since the formation of each peptide bond is accompanied by the release of a water molecule, the main goal of this contribution is to verify how the presence of H2O molecules affects the condensation of two DAA systems.
Computational methods
The structures of the reactants, transition states, intermediate and final products were optimized by employing the second–order Møller–Plesset perturbational method (MP2) [28,29,30] with the aug-cc-pVDZ basis set [31,32,33]. The analytical calculations of harmonic vibrational frequencies computed at the same level of theory allowed verification of the nature of all stationary points and thereby they were identified as either minima (no imaginary frequency) or first order saddle (one imaginary frequency). For each transition state structure, we carried out the intrinsic reaction coordinate (IRC) calculations [34,35,36] in both forward and reverse directions at the same MP2/aug-cc-pVDZ level of theory. The electronic energies of all stationary points were then refined by employing the CCSD(T) method (coupled-cluster method with single and double excitations augmented with noniterative triple excitations) [37, 38] using the same aug-cc-pVDZ basis set.
Transition state theory [39] was used to obtain rate constants (k298) for the investigated reaction (\( {k}_{298}=\raisebox{1ex}{$ kT$}\!\left/ \!\raisebox{-1ex}{$h$}\right.{\mathit{\exp}}^{\raisebox{1ex}{$-\Delta {G}^{\#}$}\!\left/ \!\raisebox{-1ex}{$ RT$}\right.} \)) where k is the Boltzmann constant, T is the temperature (298.15 K), and h stands for Planck’s constant. The Gibbs free energies (ΔG#) of the transition states were evaluated using the CCSD(T)/aug-cc-pVDZ electronic energies, while the entropy contributions, zero-point-energy corrections, and thermal corrections (at T = 298.15 K) were estimated at the MP2/aug-cc-pVDZ theory level.
All calculations were performed using the Gaussian16 program package [40], whereas the the Chemcraft program [41] was utilized to visualize the equilibrium structures of all compounds.
While performing our calculations, we assumed that the starting reactants consist of two DAA molecules in their canonical forms (i.e., containing two NH2 and two COOH groups intact) and a single water molecule.
Results and discussion
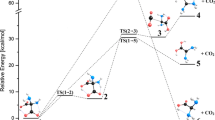
The reaction mechanisms studied in this contribution and related to the water-assisted peptide bond formation between two DAA molecules are depicted in Figs. 2, 3, 4, and 5 together with relative energies of all systems involved. As mentioned in the preceding section, the canonical forms of DAA molecules (see Fig. 1) have been chosen as the starting reactants due to their highest stability in the gas phase.

Energy profile for the water assisted peptide bond formation between two double amino acid molecules according to the concerted mechanism and leading to the trans DAA–DAA. Relative energies (in kcal mol–1) of the stationary points were obtained at the CCSD(T)/aug-cc-pVDZ//MP2/aug-cc-pVDZ level of theory (the energy of the noninteracting reacants was taken as zero)

Energy profile for the water assisted peptide bond formation between two double amino acid molecules according to the concerted mechanism and leading to the cis DAA–DAA. Relative energies (in kcal mol–1) of the stationary points were obtained at the CCSD(T)/aug-cc-pVDZ//MP2/aug-cc-pVDZ level of theory (the energy of the noninteracting reacants was taken as zero)

Energy profile for the water assisted peptide bond formation between two double amino acid molecules according to the stepwise mechanism and leading to the trans DAA–DAA. Relative energies (in kcal mol–1) of the stationary points were obtained at the CCSD(T)/aug-cc-pVDZ//MP2/aug-cc-pVDZ level of theory (the energy of the noninteracting reacants was taken as zero)

Energy profile for the water assisted peptide bond formation between two double amino acid molecules according to the stepwise mechanism and leading to the cis DAA–DAA. Relative energies (in kcal mol–1) of the stationary points were obtained at the CCSD(T)/aug-cc-pVDZ//MP2/aug-cc-pVDZ level of theory (the energy of the noninteracting reacants was taken as zero)
Concerted mechanisms
The mechanisms presented in Figs. 2 and 3 correspond to the reaction paths leading to DAA–DAA dipeptide containing its peptide bond in either trans or cis configuration, respectively. Since in both cases the reactants and products are connected by only one transition state (TS) structure, these mechanisms can be considered as concerted. The initial step in both reactions involves a formation of the reactant complex (RC) whose energy is lower than that of the noninteracting substrates by 21.2 kcal mol–1. Clearly, the RC gains its stability due to formation of intra- and inter-molecular hydrogen bonds depicted in Figs. 2 and 3. The analysis of both concerted mechanisms reveals that the peptide bond is formed through a transition state structure containing a six-membered ring involving a water molecule (unlike the highly strained four-membered ring TS structures found earlier for the corresponding noncatalyzed concerted mechanisms [27]). It is important to notice that in these two TS structures (for which the imaginary vibration frequencies are equal to 837i cm−1 and 630i cm−1 for the trans and cis route, respectively) the water molecule acts as both a hydrogen atom acceptor and donor with respect to the amino group of one DAA and the OH group of the second DAA molecule. This intermolecular hydrogen transfer where atom H2 goes to the atom O(w) and simultaneously atom H2(w) belonging to the water molecule migrates to oxygen atom O′ is accompanied by the C′–O′ bond cleavage and leads to the elimination of the H2O molecule (see the TS structures depicted in Figs. 2 and 3). The calculated energy barrier that has to be surmounted in this step for the route leading to the trans product is equal to 26.6 kcal mol–1 (the related rate constant k(ct) is equal to 5.43 × 10−3 s−1), whereas the corresponding barrier predicted for the path leading to the cis structure is much larger and equals 44.0 kcal mol–1 (the related rate constant k(cc) is equal to 2.26 × 10−15 s−1). After passing the saddle point, the final product corresponding to the DAA–DAA is formed. Depending on the mechanism considered, the resulting dipeptide contains its peptide bond in either trans or cis configuration. Since the presence of one H2O molecule (playing the catalyst role) was initially assumed and taking into account that the formation of a single peptide bond results in elimination of another H2O molecule, the interaction of such three systems has to be considered to assure consistency of the energy profile investigated. As it turns out, the lowest energy final structure (i.e., “product complex” labeled PC in Figs. 2 and 3) corresponds to the complex of two water molecules and either a trans or cis DAA–DAA isomer.
As shown in Figs. 2 and 3, the process of peptide bond formation between two DAA molecules with the assistance of a water molecule and undergoing the concerted mechanism is energetically favorable by 26.1 kcal mol–1 (when the trans DAA–DAA is formed) or 13.0 kcal mol–1 (when the cis DAA–DAA is the final product). However, one may notice that the initial reactant complex is actually lower in energy than the final complex involving cis DAA–DAA and two water molecules (see Fig. 3), hence the route leading to the cis DAA–DAA should not be considered plausible. Moreover, the height of the activation barrier for this path (44.0 kcal mol–1) also seems to be too large to render this route operative. On the other hand, the final complex of trans DAA–DAA and two water molecules (see PC in Fig. 2) represents the lowest energy structure on the corresponding energy profile as its energy is smaller than that of the initial reactant complex (RC in Fig. 2) by ca. 5 kcal mol–1. Also the height of the activation barrier for the formation of trans DAA–DAA (26.6 kcal mol–1) indicates that this path can be considered operative.
Therefore, we conclude that only the trans DAA–DAA dipeptide can be generated according to the concerted mechanism predicted for the water-assisted peptide bond formation between two DAA species due to both thermodynamical stability thereof with respect to the reactant complex (the final complex of cis DAA–DAA and two water molecules is higher in energy than the corresponding RC complex) and significantly lower activation barrier that has to be surmounted compared to the trans pathway.
Stepwise mechanisms
Our calculations indicate that the water-assisted DAA–DAA dipeptide formation may alternatively proceed according to the stepwise mechanism. The energy profiles corresponding to the reactions leading to trans and cis dipeptide are shown in Figs. 4 and 5, respectively. The initial step in both processes is the formation of the hydrogen bonded reactant complex (RC). It should be noted, however, that the RC structure presented in Figs. 4 and 5 (i.e., for the stepwise route) is actually different than the RC shown in Figs. 2 and 3 (i.e., for the concerted route) because in both cases the RC local minimum energy structure was found by employing the IRC calculations and matches its corresponding transition state as such. In particular, the reactant complex predicted for the stepwise mechanisms consists of two DAA amino acids connected together via the H-bonds and H2O molecule whose oxygen and hydrogen atoms form hydrogen bonds with functional groups of both DAAs (see RC structure in Figs. 4 and 5). The resulting H-bond network renders this reactant complex stable with respect to the noninteracting substrates by 7.7 kcal mol–1. In the next step, the RC passes through the energy barrier of 9.2 kcal mol–1 (the corresponding rate constant k(s1) is equal to 2.13 × 109 s−1) via the transition state structure (labeled TS1 in Figs. 4 and 5) to form the intermediate product (Int). The analysis of both the saddle point structure and the imaginary vibration mode (υ = 1383i cm−1) reveals that the peptide C′–N bond is formed during this step by the nucleophilic attack of the N atom of the NH2 group of one double amino acid molecule to the C′ atom of the carboxylic group available in the second DAA. In addition, the hydrogen H2 migrates from the NH2 group to the oxygen atom of the water molecule O(w) with simultaneous transfer of the second hydrogen atom H2(w) from the water molecule to O′ oxygen atom belonging to the second DAA. The energy profiles show that the resulting intermediate product (Int) is more stable than the reactants by 20.1 kcal mol–1. It is also important to emphasize that the C′–N bond is already formed in the Int structure.
Once this intermediate product structure is achieved, the two routes become possible, leading either to the trans DAA–DAA or to the cis DAA–DAA product. The reaction resulting in the formation of trans DAA–DAA requires passing the energy barrier of 23.4 kcal mol–1 and proceeds through the TS2 transition state (the corresponding rate constant k(s2t) is equal to 2.6 s−1). Analysis of the imaginary frequency mode (596i cm−1) shows that during this process, the hydrogen atom migrates (through the assisting water molecule) between two OH groups connected to the same carbon atom in the Int structure, namely the H2’ atom goes to the O(w) oxygen atom and the H2(w) hydrogen atom transfers to the O1’ oxygen. This process is accompanied by the C′–O1’ bond cleavage and elimination of another H2O system, see the TS2 and Int structures depicted in Fig. 4. The final product (PC) formed along this path represents the trans DAA–DAA dipeptide interacting with the water dimer and the energy of this complex is lower than that of the starting reactants by 26.4 kcal mol–1. Alternatively, the intermediate product Int might evolve to the DAA–DAA containing its peptide bond in the cis configuration, see Fig. 5. Although the energy barrier that has to be overcome in that case is equal to 25.7 kcal mol–1 (which leads to the rate constant k(s2c) of 6.75 s−1) and was predicted to be slightly higher than that found for the route leading to the trans DAA–DAA, the analysis of the TS2 transition state structure and the imaginary vibration mode (1026i cm−1) leads to similar conclusions. Namely, the hydrogen atom H1’ is being transferred to the assisting water molecule with simultaneous migration of the H2(w) atom to the O2’ oxygen, which is accompanied by the C′–O2’ bond rupture and elimination of another H2O, see Fig. 5. In fact, the TS2 structures predicted for the paths leading to trans and cis dipeptide and depicted in Figs. 4 and 5 differ only by mutual orientation of two amino acids and the water molecule, which enables the formation of the DAA–DAA containing its peptide bond in various configurations. The final product on the path leading to the cis DAA–DAA represents the complex of the dipeptide interacting with two water molecules, and its energy is lower by 13.7 kcal mol–1 than the energy of the reactants.
The comparison of the energy profiles predicted for two stepwise mechanisms and given in Figs. 4 and 5 reveals that the energy barriers that have to be surmounted are similar for both paths (although the second barrier is slightly smaller for the route leading to the trans DAA–DAA), whereas the energies of the final product complexes differ significantly (i.e., the PC including trans dipeptide is more stable by 12.7 kcal mol–1 than that including the cis dipeptide). Clearly, this difference is mainly caused by the larger stability of trans in comparison to cis DAA–DAA isomer and also by different H-bond networks formed by each of these structures with two water molecules.
Thus, we conclude that the water-assisted peptide bond formation between two DAA molecules according to the stepwise mechanisms should lead to both trans and cis isomers of DAA–DAA; however, the former isomer is expected to dominate in the bulk.
Conclusions
The formation of trans and cis peptide bonds (C–N) between two double amino acids catalyzed by a single H2O molecule and proceeding along either concerted or stepwise paths have been studied at the CCSD(T)/aug-cc-pVDZ//MP2/aug-cc-pVDZ level of theory. The results obtained for the concerted mechanisms reveal that only the path leading to the trans DAA–DAA should be considered operative, whereas a relatively large energy barrier and large stability of the initial complex render the formation of cis DAA–DAA not plausible. On the other hand, the formation of both trans and cis isomers seems likely when the stepwise paths are concerned. In any case, the trans DAA–DAA isomers are expected to dominate in the final product due to the smaller energy barriers on the paths leading to these conformers and because of larger stability of the trans DAA–DAA structure. Recalling that the trans conformation of the peptide bond is typically favored in most peptide structures due to the smaller steric repulsion between the two Cα atoms (the Cα–C–N–Cα dihedral angle approaches 180° in trans and 0° in cis conformation), whereas the cis peptide bond occurs more frequently only when the proline residue is involved [42,43,44], we conclude that the peptide bond of trans configurations should also be preferred when formed by DAAs.
The presence of a water molecule during the formation of DAA–DAA dipeptide has been proven important regardless of the conformation of the final product as the energy barriers predicted for the water-assisted mechanisms are significantly reduced in comparison to the barriers found for the corresponding uncatalyzed reactions [27]. In order to explain the catalytic role of the H2O molecule in this process, one may follow the speculation provided in ref. [45] based on the comparison of the stability of four- vs six-membered rings. Namely, the steric strain existing in the four-membered ring transition state structures (that are characterized in the uncatalyzed mechanisms) likely raises the energies of those systems. By contrast, in the water assisted mechanisms, the transition state structures involve six-membered rings with smaller strain and thus higher stability.
References
Oie T, Loew GH, Burt SK, Binkley JS, MacElroy RD (1982) J Am Chem Soc 104(23):6169–6174. https://doi.org/10.1021/ja00387a001
Jensen JH, Baldridge KK, Gordon MS (1992) J Phys Chem 96(21):8340–8351. https://doi.org/10.1021/j100200a025
Redondo P, Martínez H, Cimas Á, Barrientos C, Largo A (2013) Phys Chem Chem Phys 15(31):13005–13012. https://doi.org/10.1039/c3cp51535d
Van Dornshuld E, Vergenz RA, Tschumper GS (2014) J Phys Chem B 118(29):8583–8590. https://doi.org/10.1021/jp504924c
Montalbetti CAGN, Falque V (2005) Tetrahedron 61(46):10827–10852. https://doi.org/10.1016/j.tet.2005.08.031
Berg J, Tymoczko J, Stryer L (2002) Bicochemistry. Freeman, New York
Georgelin T, Jaber M, Bazzi H, Lambert JF (2013) Orig Life Evol Biosph 43(4–5):429–443. https://doi.org/10.1007/s11084-013-9345-2
Shanker U, Bhushan B, Bhattacharjee G, Kamaluddin (2012) Orig Life Evol Biosph 42(1):31–45. https://doi.org/10.1007/s11084-012-9266-5
Rimola A, Tosoni S, Sodupe M, Ugliengo P (2006) ChemPhysChem 7(1):157–163. https://doi.org/10.1002/cphc.200500401
Basiuk VA, Sainz-Rojas J (2001) Adv Sp Res 27(2):225–230. https://doi.org/10.1016/S0273-1177(01)00051-5
Bujdák J, Rode BMJ (2002) Inorg Biochem 90(1–2):1–7. https://doi.org/10.1016/S0162-0134(02)00395-1
Oie T, Loew GH, Burt SK, MacElroy RD (1984) J Am Chem Soc 106(26):8007–8013. https://doi.org/10.1021/ja00338a001
Yamagata Y, Inomata K (1997) Orig Life Evol Biosph 27(4):339–344. https://doi.org/10.1023/A:1006529421813
Rode BM, Suwannachot Y (1999) Coord Chem Rev 190–192:1085–1099. https://doi.org/10.1016/S0010-8545(99)00159-9
Rimola A, Rodríguez-Santiago L, Ugliengo P, Sodupe M (2007) J Phys Chem B 111(20):5740–5747. https://doi.org/10.1021/jp071071o
Remko M, Michael B (2001) Phys Chem Chem Phys 3:4667–4673. https://doi.org/10.1039/b105623a
Oie T, Loew GH, Burt SK, Binkley JS, MacElroy RD (1982) Int J Quantum Chem 22(9 S):223–245. https://doi.org/10.1002/qua.560220722
Thirumoorthy K, Nandi N (2007) J Mol Struct Theochem 818(1–3):107–118. https://doi.org/10.1016/j.theochem.2007.05.017
Oie T, Loew GH, Burt SK, MacElroy RD (1983) J Am Chem Soc 105(8):2221–2227. https://doi.org/10.1021/ja00346a020
Rimola A, Tosoni S, Sodupe M, Ugliengo P (2005) Chem Phys Lett 408(4–6):295–301. https://doi.org/10.1016/j.cplett.2005.04.056
Li Y, Li F, Zhu Y, Li X, Zhou Z, Liu C, Zhang W, Tang M (2016) Struct Chem 27(4):1165–1173. https://doi.org/10.1007/s11224-016-0740-y
Williams IH (1987) J Am Chem Soc. https://doi.org/10.1021/ja00255a012
Freza S, Marchaj M, Skurski P (2014) Chem Phys Lett 599:34–37. https://doi.org/10.1016/j.cplett.2014.03.020
Freza S (2017) Theor Chem Accounts 136:7. https://doi.org/10.1007/s00214-016-2031-5
Freza S (2016) Theor Chem Accounts 135:146. https://doi.org/10.1007/s00214-016-1906-9
Freza S (2018) Struct Chem 29(4):1025–1029. https://doi.org/10.1007/s11224-018-1085-5
Czapla M, Freza S (2017) Int J Quantum Chem 117(21):e25435. https://doi.org/10.1002/qua.25435
Møller C, Plesset MS (1934) Phys Rev 46(7):618–622. https://doi.org/10.1103/PhysRev.46.618
Head-Gordon M, Pople JA, Frisch M (1988) J Chem Phys Lett 153(6):503–506. https://doi.org/10.1016/0009-2614(88)85250-3
Frisch MJ, Head-Gordon M, Pople JA (1990) Chem Phys Lett 166(3):275–280. https://doi.org/10.1016/0009-2614(90)80029-D
Dunning Jr THJ (1989) Chem Phys. https://doi.org/10.1063/1.456153
Kendall RA, Dunning TH, Harrison RJJ (1992) Chem Phys 96(9):6796–6806. https://doi.org/10.1063/1.462569
Woon DE, Dunning THJ (1993) Chem Phys 98(2):1358–1371. https://doi.org/10.1063/1.464303
Hratchian HP, Schlegel HBJ (2004) Chem Phys 120(21):9918–9924. https://doi.org/10.1063/1.1724823
Hratchian HP, Schlegel HB (2005) J Chem Theory Comput 1(1):61–69. https://doi.org/10.1021/ct0499783
Fukui K (1981) Acc Chem Res 14(12):363–368. https://doi.org/10.1021/ar00072a001
Purvis GD, Bartlett RJJ (1982) Chem Phys 76(4):1910–1918. https://doi.org/10.1063/1.443164
Bartlett RJ, Purvis GD (1978) Int J Quantum Chem 14(5):561–581. https://doi.org/10.1002/qua.560140504
Lowry TH, Richardson K (1981) Mechanism and theory in organic chemistry, 2nd edn. Harper & Row, New York
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H et al. (2016) Gaussian 16, revision B.01. Gaussian Inc, Wallingford
Chemcraft- graphical software for visualization of quantum chemistry computations. https://www.chemcraftprog.com
Grathwohl C, Wüthrich K (1976) Biopolymers 15(10):2025–2041. https://doi.org/10.1002/bip.1976.360151012
Brandts JF, Halvorson HR, Brennan M (1975) Biochemistry 14(22):4953–4963. https://doi.org/10.1021/bi00693a026
Juy M, Lam-Thanh H, Lintner K, Fermandjian S (1983) Int J Pept Protein Res 22(4):437–449. https://doi.org/10.1111/j.1399-3011.1983.tb02113.x
Carey FA, Sundberg RJ (2007) Advanced organic chemistry part A. https://doi.org/10.1021/ed065pA139.2
Acknowledgments
This research was supported by the Polish Ministry of Science and Higher Education grant No. DS 530-8375-D499-19. The calculations have been carried out using resources provided by Wroclaw Centre for Networking and Supercomputing (http://wcss.pl) grant No. 436.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Freza, S. Water-assisted peptide bond formation between two double amino acid molecules in the gas phase. J Mol Model 25, 184 (2019). https://doi.org/10.1007/s00894-019-4081-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-4081-9