
Abstract
Cyclic organic peroxides are a broad and highly sought-after class of peroxide compounds that present high reactivity and even explosive character. The unusually high reactivity of these peroxides can generally be attributed to the rupture of O–O bonds. Cyclic diperoxides are a very interesting series of substituted compounds in which tetroxane is the most prominent member. Gas-phase thermolysis of the simplest substituted member of the series [3-methyl-1,2,4,5-tetroxane or methylformaldehyde diperoxide (MFDP)] has been observed to yield one acetaldehyde, one formaldehyde, and one oxygen molecule as reaction products. DFT at the 6-311 + G** level of theory using the BHANDHLYP correlation–exchange functional was applied via the Gaussian09 program to calculate the critical points of the potential energy surface (PES) of this reaction. Equatorial and axial isomers were studied. The singlet state PES of MFDP was calculated, and an open diradical structure was found to be the first intermediate in a stepwise reaction. Two PESs were subsequently obtained: singlet state (S) and triplet state (T) PESs. After that, two alternative stepwise reactions were found to be possible: 1) one in which either an acetaldehyde, or 2) formaldehyde molecule is initially formed. For second one, exothermic reactions were observed for both the S and T PESs. The reaction products include a oxygen molecule in either S or T state, with the T reaction being the most exothermic. When calculations were performed at the CASSCF(10,10)/6-311 + G** level, spin–orbit coupling permitted S to T crossing at the open diradical intermediate stage, a non-adiabatic reaction was observed, and lower activation energies and higher exothermicity were generally seen for the T PES than for the S PES. These results were compared with the corresponding results for tetroxane. The spin–orbit coupling of MFDP and tetroxane yielded identical values, so it appears that the methyl substituent does not have any effect on this coupling.











Similar content being viewed by others

References
Jones CW (1999) Applications of hydrogen peroxides and derivatives. Royal Society of Chemistry, Cambridge
Jones CW, Patai S (1983) The chemistry of peroxides. Wiley, New York
Ando W (1992) Organic peroxides. Wiley, New York
Adam W (2000) Peroxide chemistry: mechanistic and preparative aspects of oxygen transfer. Wiley-VCH, Weinheim
Oxley JC, Smith JL, Chen H (2002) Decomposition of a multi-peroxidic compound: triacetone triperoxide (TATP). Propellants Explos Pyrotech 27:209–216
Vennerstrom JL, Ager AL, Andersen SL, Grace JM, Wongpanich V, Angerhofer CK (2000) Assessment of the antimalarial potential of tetraoxane. Am J Trop Med Hyg 62(5):573–578
Li Y, Zhu H, Jiang J, Pan JP, Wu GS, Wu JM, Shi YL, Yang JD, Wu BA (2000) Synthesis and antimalarial activity of artemisinin derivatives containing an amino group. J Med Chem 43:1635–1640
Wang X, Dong Y, Wittlin S, Charman SA, Chiu FC, Chollet J, Katneni K, Mannila J, Morizzi J, Ryan E, Scheurer C, Steuten J, Santo Tomas J, Snyder C, Vennerstrom JL (2013) Comparative antimalarial activities and ADME profiles of ozonides (1,2,4-trioxolanes) OZ277, OZ439, and their 1,2-dioxolane, 1,2,4-trioxane, and 1,2,4,5-tetraoxane isosteres. J Med Chem 56(6):2547–2555
Kumar N, Singh R, Rawat DS (2012) Tetraoxanes: synthetic and medicinal chemistry perspective. Med Res Rev 32(3):581–610
Franco LL, De Almeida MV, Silva LFE, Vieira PP, Pohlit AM, Valle MS (2012) Synthesis and antimalarial activity of dihydroperoxides and tetraoxanes conjugated with bis(benzyl)acetone derivatives. Chem Biol Drug Des 79(5):790–797
Leiva LCA, Jorge NL, Romero JM, Cafferata LFR, Gómez Vara ME, Castro EA (2008) Decomposition of the acetone cyclic diperoxide in octanol solution. J Argent Chem Soc 96(1–2):110–122
Profeta MI, Romero JM, Leiva LCA, Jorge NL, Gómez Vara ME, Castro EA (2011) Solvent effect of oxygen in the thermolisys decomposition of the acetone diperoxide. Meth Appl Chemoinf Chem Eng 96(1–2):110–122
Pila AN, Profeta MI, Romero JM, Jorge NL, Castro EA (2012) Kinetics and mechanism of the thermal decomposition reaction of 3,6-diphenyl-1,2,3,5-tetroxane in solution. Int J Chem Model 4(4):5–10
Reguera MB, Frette SG, Romero JM, Jorge NL, Castro EA (2012) Synthesis and thermical decomposition reaction of 3,6-dibutanoic-1,2,4,5-tetroxane in solution. Bentham Sci Newsl 4(1):1–4
Jorge NL, Romero JM, Grand A, Hernández-Laguna A (2012) Gas-phase thermolysis reaction of formaldehyde diperoxide. Kinetic study and theoretical mechanisms. Chem Phys 39:37–45
Reguero M, Bernardi F, Bottoni A, Olivucci M, Robb MA (1991) Chemiluminescent decomposition of 1,2-dioxetanes: an MC-SCF-MP2 study with VB analysis. J Am Chem Soc 113:1566–1572
Wilsey S, Bernardi F, Olivucci M, Robb M, Murphy S, Adam W (1999) The thermal decomposition of 1,2-dioxetane revisited. J Phys Chem 103:1669–1677
Tanaka S, Tanaka J (2000) Ab initio molecular orbital studies on the chemiluminescence of 1,2-dioxetanes. J Phys Chem A 104(10):2078–2090
Cafferata LFR, Lombardo JD (1994) Kinetic and mechanism of the thermal decomposition reaction of acetone cyclic diperoxide in the gas phase. Int J Chem Kinet 26:503–509
Cazut SAI, Ramírez Maisuls EH, Delfino MR, Romero JM, Jorge NL, Castro EA (2009) Thermal decomposition of formaldehyde diperoxide in aqueous solution. Russ J Gen Chem 79(10):2187–2190
Leiva LCA, Jorge NL, Romero JM, Cafferata LFR, Gómez Vara ME, Castro EA (2010) Thermal decomposition reaction of 3,3,6,6-tetramethyl- 1,2,4,5-tetroxane in 2-methoxy-ethanol solution. Chem Heterocycl Compd 45(12):1455–1459
Leiva LCA, Castellanos GB, Jorge NL, Cafferata LFR, Gómez Vara ME (1998) Kinetics and mechanism of the thermal decomposition reaction of 3,3,6,6-tetramethyl-1,2,4,5-tetroxane in methanol solution. Rev Soc Quím Méx 42(5):223–227
Barreto GP, Eyler GN (2011) Thermal decomposition of 3,3,6,6,9,9-hexaethyl-1,2,4,5,7,8-hexaoxacyclononane in solution and its use in methyl methacrylate polymerization. Polym Bull 67(1):1–14
Iglesias M, Barreto GP, Eyler GN, Cañizo AI (2010) Thermal decomposition of cyclic organic peroxides in pure solvents and binary solvent mixtures. Int J Chem Kinet 42(6):347–353
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38:3098–3100
Slater JC (1974) Quantum theory of molecules and solids, the self-consistent field for molecules and solids, vol 4. McGraw-Hill, New York
Lee C, Yang LW, Par RG (1988) Development of the Colle–Salvetti conelation energy formula into a functional of the electron density. Phys Rev B 37:785–789
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA et al (2004) Gaussian 03, revision C.01. Gaussian Inc., Wallingford
Abegg PW, Ha TK (1974) Ab initio calculation of spin-orbit-coupling constant from Gaussian lobe SCF molecular wavefunctions. Mol Phys 27:763–767
Abegg PW (1975) Ab initio calculation of spin-orbit-coupling constants for Gaussian lobe and Gaussian-type wave-functions. Mol Phys 30:579–596
Flúkiger P, Lúthi HP, Portmann S, Weber J (2000) Gaussview 5.0. Swiss Center for Scientific Computing, Manno
Gonzalez C, Schlegel HB (1989) An improved algorithm for reaction path following. J Chem Phys 90:2154–2161
Gonzalez C, Schlegel HB (1990) Reaction path following in mass-weighted internal coordinates. J Phys Chem 94:5523–5527
Eade RHE, Robb MA (1981) Direct minimization in MC SCF theory: the quasi-Newton method. Chem Phys Lett 83:362–368
Siegbahn EM (1984) A new direct CI method for large CI expansions in a small orbital space. Chem Phys Lett 109:417–423
Bernardi F, Olivucci M, Robb MA (1990) Mechanism of ground-state-forbidden photochemical pericyclic reactions: evidence for real conical intersections. J Am Chem Soc 112:1737–1744
Klene M, Robb MA, Frisch MJ, Celani P (2000) Parallel implementation of the CI-vector evaluation in full CI/CAS-SCF. J Chem Phys 113:5653–5665
Koseki S, Gordon MS, Schmidt MW, Matsunaga N (1995) Main-group effective nuclear charges for spin-orbit calculations. J Phys Chem 99:12764–12772
Koseki SS, Schmidt MW, Gordon MS (1998) Effective nuclear charges for the first- through third-row transition metal elements in spin-orbit calculations. J Phys Chem 102:10430–10435
Leffler JE (1953) Parameters for the description of transition states. Science 117:340–341
Hammond GS (1955) A correlation of reaction rate. J Am Chem Soc 77:334–338
Mathieu D (2012) Theoretical shock sensitivity index for explosives. J Phys Chem A 116(7):1794–1800
Mathieu D (2013) Toward a physically based quantitative modeling of impact sensitivities. J Phys Chem A 117(10):2253–2259
Yu Z, Bernstein ER (2013) Sensitivity and performance of azole-based energetic materials. J Phys Chem A 117(42):10889–10902
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper belongs to Topical Collection QUITEL 2013
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure S1
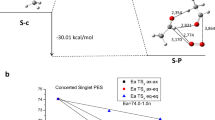
Reaction profile of pathway S-Ia in Scheme 1 for the axial isomer. Activation energies are calculated from S-c or S-o. Energies are in kcal/mol, and bond lengths are in Å. Spin densities are shown next to the respective atoms in parentheses. White, gray, and red spheres indicate hydrogen, carbon, and oxygen atoms, respectively. (DOCX 294 kb)
Figure S2
Reaction profile of pathway S-Ib in Scheme 1 for the axial isomer. Activation energies are calculated from S-c or S-o. Energies are in kcal/mol, and bond lengths are in Å. Spin densities are shown next to the respective atoms in parentheses. White, gray, and red spheres indicate hydrogen, carbon, and oxygen atoms, respectively. (DOCX 283 kb)
Figure S3
Reaction profile of the second part of pathway S-Ia in Scheme 1 for the axial isomer. Energies are in kcal/mol, and bond lengths are in Å. Spin densities are shown next to the respective atoms in parentheses. White, gray and red spheres indicate hydrogen, carbon, and oxygen atoms, respectively. (DOCX 102 kb)
Figure S4
Reaction profile of pathway S-II in Scheme 1 for the axial isomer. Energies are in kcal/mol, and bond lengths are in Å. White, gray, and red spheres indicate hydrogen, carbon, and oxygen atoms, respectively. (DOCX 115 kb)
Figure S5
Reaction profile of the first step of pathway T-Ia in Scheme 2 for the axial isomer. Activation energies are calculated from T-o. Energies are in kcal/mol, and bond lengths are in Å. Spin densities are shown next to the respective atoms in parentheses. White, gray, and red spheres indicate hydrogen, carbon, and oxygen atoms, respectively. (DOCX 138 kb)
Figure S6
Reaction profile of the first step of pathway T-Ib in Scheme 2 for the axial isomer. Activation energies are calculated from T-o. Energies are in kcal/mol, and bond lengths are in Å. Spin densities are shown next to the respective atoms in parentheses. White, gray, and red spheres indicate hydrogen, carbon, and oxygen atoms, respectively. (DOCX 134 kb)
Rights and permissions
About this article

Cite this article
Profeta, M.I., Romero, J.M., Jorge, N.L. et al. Theoretical study of the gas-phase thermolysis of 3-methyl-1,2,4,5-tetroxane. J Mol Model 20, 2224 (2014). https://doi.org/10.1007/s00894-014-2224-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-014-2224-6