
Abstract
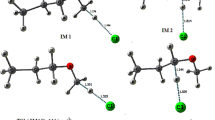
The present work deals with the theoretical investigation on the Cl initiated H-atom abstraction reaction of sevoflurane, (CF3)2CHOCH2F. A dual-level procedure has been adopted for studying the kinetics of the reaction. Geometrical optimization and frequency calculation were performed at DFT(BHandHLYP)/6-311G(d,p) while single-point energy calculation was made at CCSD(T)/6-311G(d,p) level of theory. The intrinsic reaction coordinate (IRC) calculation has also been performed to confirm the smooth transition from the reactant to product through the respective transition state. The rate constants were calculated using conventional transition state theory (TST). It has been found that 99 % of the reaction proceeded via the H-atom abstraction from the –CH2F end of the sevoflurane. The rate constant of the dominant path is found to be 1.13 × 10−13 cm3 molecule−1 s−1. This is in excellent agreement with the reported experimental rate constant of 1.10 × 10−13 cm3 molecule−1 s−1 obtained by relative rate method using FTIR/Smog chamber and LP/LIF techniques.



Similar content being viewed by others
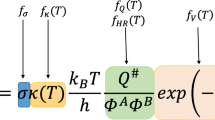

References
Guenther A, Hewitt CN, Erickson D, Fall R, Geron C, Graedel T, Harley P, Klinger L, Lerdau M, McKay WA, Pierce T, Scholes B, Steinbrecher R, Tallamraju R, Taylor J, Zimmermann P (1995) J Geophys Res 100:8873–8892
Sawyer RF, Harley RA, Cadle SH, Norbeck JM, Slott R, Bravo HA (2000) Atoms Environ 34:2161–2181
Placet M, Mann CO, Gilbert RO, Niefer MJ (2000) Atoms Environ 34:2183–2204
Guenther A, Geron C, Pierce T, Lamb B, Harley P, Fall R (2000) Atoms Environ 34:2205–2230
Atkinson R, Arey J (2003) Chem Rev 103:4605–4638
Sulbaek Andersen MP, Sander SP, Nielsen OJ, Wagner DS, Sanford TJ Jr, Wallington TJ (2010) Br J Anaesth 105:760–766
Wallington TJ, Hurley MD, Fedotov V, Morrell C, Hancock G (2002) J Phys Chem A 106:8391–8398
Sulbaek Andersen MP, Nielsen OJ, Karpichev B, Wallington TJ, Sander SP (2012) J Phys Chem A 116:5806–5820
Dalmasso P, Taccone R, Nieto J, Teruel M, Lane S (2006) Atmos Environ 40:7298–7307
Wingenter OW, Kubo MK, Blake NJ, Smith TW, Blake DR, Rowland FS (1996) J Geophys Res 101:4331–4340
Freitas MP, Buhl M, Hagan DO, Cormanich RA, Tormena CF (2012) J Phys Chem A 116:1677–1682
Lesarri A, Vega-Toribio A, Suenram RD, Brugh DJ, Grabow JU (2010) Phys Chem Chem Phys 12:9624–9631
Tang P, Zubryzcki I, Xu Y (2001) J Comput Chem 22:436–444
Truhlar DG (1995) In: Heidrich D (ed) The reaction path in chemistry: current approaches and perspectives. Kluwer, Dordrecht
Hu WP, Truhlar DG (1996) J Am Chem Soc 118:860–869
Truhlar DG, Garrett BC, Klippenstein SJ (1996) J Phys Chem A 100:12771–12800
Becke AD (1993) J Chem Phys 98:1372–1377
Pople JA, Head-Gordon M, Raghavachari K (1987) J Chem Phys 87:5968–5975
Frisch MJ et al. (2010) Gaussian 09 (Version C.01). Gaussian Inc, Wallingford
Peng C, Schlegel HB (1993) Isr J Chem 33:449
Peng C, Ayala PY, Schlegel HB, Frisch MJ (1996) J Comput Chem 17:49–56
Gonzales C, Schlegel HB (1990) J Chem Phys 94:5523–5527
Gonzales C, Schlegel HB (1991) J Chem Phys 95:5853–5860
Wu Q, Zhu W, Xiao H (2013) J Mol Model 19:2945–2954
Lide DR (ed) (1994) CRC Handbook of Chemistry and Physics, 75th edn. CRC, New York
Fischer H, Radom L (2001) Angew Chemie Int Ed 40:1340–1371
Sun H, Gong H, Pan X, Hao L, Sun CC, Wang R, Huang X (2009) J Phys Chem A 113:5951–5957
Song G, Jia X, Gao Y, Luo J, Yu Y, Wang R, Pan X (2010) J Phys Chem A 114:9057–9068
Johnson RD (ed) (2011) NIST Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database 101, Release 15b, August 2011. http://cccbdb.nist.gov/
Frisch A, Nielsen AB, Holder AJ et al. (2009) Gauss-View 05. Gaussian Inc, Wallingford, CT
Laidler KJ (2004) Chemical Kinetics, 3rd edn. Pearson Education, New Delhi
Truhlar DG, Garrett BC, Klippenstein SJ (1996) J Phys Chem 100:12771–12800
Wigner EP (1932) Z Phys Chem B19:203–216
Chase MW Jr, Davies CA, Downey JR Jr, Frurip DJ, McDonald RA, Syverud AN (1985) JANAF thermochemical tables. J Phys Chem Ref Data 14: Suppl 1, 3rd edn
Acknowledgments
Authors are thankful to Council of Scientific and Industrial Research (CSIR), New Delhi for providing financial assistance during the course of the present investigation. PKR is thankful to University Grants Commission, New Delhi for providing Rajiv Gandhi National Fellowship (RGNF). Authors are also thankful to UP State Government for providing a grant under its Center of Excellence program and to UGC under SAP program for establishing the computational lab.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Singh, H.J., Gour, N.K., Rao, P.K. et al. Theoretical investigation on the kinetics and branching ratio of the gas phase reaction of sevoflurane with Cl atom. J Mol Model 19, 4815–4822 (2013). https://doi.org/10.1007/s00894-013-1977-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-013-1977-7