
Abstract
Cyclophilin A has attracted attention recently as a new target of anti-human immunodeficiency virus type 1 (HIV-1) drugs. However, so far no drug against HIV-1 infection exhibiting this mechanism of action has been approved. To identify new potent candidates for inhibitors, we performed in silico screening of a commercial database of more than 1,300 drug-like compounds by using receptor-based docking studies. The candidates selected from docking studies were subsequently tested using biological assays to assess anti-HIV activities. As a result, two compounds were identified as the most active. Specifically, both exhibited anti-HIV activity against viral replication at a low concentration and relatively low cytotoxicity at the effective concentration inhibiting viral growth by 50 %. Further modification of these molecules may lead to the elucidation of potent inhibitors of HIV-1.

Docking poses of two compounds (23 and 12) with anti-HIV activity
Similar content being viewed by others

Introduction
Cyclophilin A (CypA) was discovered originally as the receptor of the immunosuppressive drug cyclosporin A (CsA)—a molecule exhibiting multiple biological functions. The formation of complexes between CypA and CsA, which is an 11-mer cyclic peptide isolated from the fungus Tolypocladium inflatum, allows CypA to interact with calcineurin, reduce the production of interferon γ and interleukin-2, and exert immunosuppressive effects [1]. CypA is one of 15 known human cyclophilins, and can catalyze cis–trans isomerization in peptide bonds containing proline via its peptidyl prolyl isomerase (PPIase) activity. Recently, it was reported that CypA interacts with the NS5A and NS5B parts of hepatitis C virus polymerase [2, 3], the nucleocapsid protein of the SARS coronavirus [4], and the capsid (CA) protein of human immunodeficiency virus type 1 (HIV-1) [5]. These multiple functions make CypA an attractive target for drug development.
In addition to CsA, the natural product sanglifehrin A [6] and several peptide analogs [7] were reported to be active inhibitors of CypA. Recently, to decrease HIV-1 infectivity by disrupting the interaction of CypA with CA, several small molecules have been developed [8, 9]. However, new drugs against HIV-1 infection exhibiting this mechanism of action have not yet been approved.
Nevertheless, there are several published reports of structural information regarding to the binding modes of CypA and CA fragments [10], CsA [11], or small peptides [12], and the available information suggests the potential applicability of in silico inhibitor screening and design for elucidating potential inhibitors. Here, we screened a small database of 1,377 small-molecular-weight compounds. A total of 29 compounds was selected according to docking scores. Together with two commercial positive control compounds, these 29 selected compounds were tested using biological assays. Finally, two compounds were identified as potent anti-HIV candidates, displaying acceptable cellular toxicity.
Materials and methods
Small molecular database for virtual screening
The structures of 1,377 low-molecular-weight compounds in a chemical structure data (SD) file format were obtained from ChemGenesis (http://www.all-chemy.com/), who supplied all the compound samples in the file. This SD file was converted into Molecular Operating Environment (MOE; Chemical Computing Group, Montreal, Canada) database format, and energy optimization of each molecule was performed under the MMFF94x forcefield using the MOE software package. The descriptors of molecular weight, lip_violation, lip_druglike [13], and diameter were calculated and typed into the fields of the database to confirm the basic properties of the compounds in the database.
Preparation of ligands and protein
The 1.64-Å resolution crystallographic data of CypA in complex with the dipeptide alanine-proline (AP; PDB entry 2CYH) [12] was obtained from the Brookhaven Protein Data Bank (http://www.pdb.org/pdb). This co-crystallized structure was used in preliminary studies to determine whether the docking parameters used were appropriate, and the structure of CypA within the structure was used as a receptor for screening the Allchemy database. In the present study, the “MOE dock” program was used to perform all the screening procedures. The Protonate3D module in MOE was used to assign ionization states, and to position hydrogen atoms into the receptor molecule. Subsequently, after adding partial charges under the MMFF94x forcefield and fixing the backbone atoms, energy minimization of the receptor was performed.
An active site covering the entire area of the two pockets in the CypA molecule was found using the SiteFinder module in MOE, and dummy atoms with hydrophobic or hydrophilic properties were placed into the two pockets to define the site.
Molecular docking
We docked AP back into the receptor (self-docking or re-docking) and screened the prepared database. For simplicity, flexibility of all of ligand atoms was allowed, in contrast to the receptor atoms during docking studies. We used five stages of the MOE Dock module to determine the potential docking poses of each ligand: (1) Dock was used to generate conformations from a single 3D conformer by applying a collection of preferred torsion angles to the rotatable bonds. Here, a systematic search was conducted covering all combinations of angles on a grid if this resulted in fewer than 5,000 conformers. Otherwise, a stochastic sampling of conformations was conducted. (2) The collection of poses was generated from the pool of ligand conformations using the Triangle Matcher method, which can align ligand triplets of atoms on triplets of alpha spheres in a relatively systematic way. (3) Poses generated by the placement methodology were rescored using the London dG scoring function, and low scores were assigned to good poses. The top 30 poses were kept. (4) These 30 poses were refined using the explicit molecular mechanics forcefield method; at this time, the forcefield was set to MMFF94x. (5) Poses resulting from the refinement stage were rescored using the London dG scoring function. The top 30 poses of each compound were retained for manual examination. Finally, candidates used for examination were selected according to the best docking score of each compound.
Inhibition of HIV-1 replication
Compounds were obtained from ChemGenesis, and positive control compounds were purchased from Namiki Shoji (http://www.namiki-s.co.jp/). Biological assays were performed to determine whether the compounds exert inhibitory effects on a single replication cycle of HIV-1. Briefly, 293T cells (1.5 × 106 cells in a 100-mm dish) were transfected with 8.9 μg of the pNL4-3-based [14], envelope glycoprotein-deficient HIV-1 proviral construct carrying a luciferase reporter gene, pNL-Luc-E-R+ [15], together with 1.1 μg of a vesicular stomatitis virus G protein (VSVG) expression plasmid, pHit/G [16], using FuGENE HD transfection reagent (Roche, Basel, Switzerland) to generate VSVG-pseudotyped HIV-1. Eighteen hours later, the transfected 293T cells were trypsinized and split into 200-μl subcultures in a 96-well plate in the presence of the indicated concentrations of a test compound. After 30 h of incubation, the cell culture supernatant was used to infect subconfluent U87.CD4.CXCR4 [17] or MT-4 cells treated with the corresponding compound. Twenty-four hours after infection, the luciferase activity in infected cells was measured using the Steady Glo Luciferase assay kit (Promega, Madison, WI) with a microplate luminometer (LB960, Berthold, Bad Wildbad, Germany) according to the manufacturer’s protocol. The inhibitory effect of the compounds on viral replication was evaluated as the reduction in luciferase activity in infected cells. In addition, a cell toxicity test was performed using the WST-1 cell proliferation assay system (Roche) according to the manufacturer’s protocol. Briefly, subconfluent 293T, U87.CD4.CXCR4, and MT-4 cells were treated with the indicated concentrations of a compound for 24 h. WST-1 reagent was then added to the cell culture, which was further incubated until the absorbance of the samples at 450 nm was approximately 1.4 (approximately 1 h for 293T and U87.CD4.CXCR4 cells and 3 h for MT-4). The absorbance of the samples was measured using a microplate reader (Multiskan FC, Thermo Scientific, Rockford, IL), and the cytotoxicity of the compounds was evaluated as a reduction in absorbance. U87.CD4.CXCR4 cells were obtained through the AIDS Research and Reference Reagent Program (Division of AIDS, NIAID, NIH) from HongKui Deng and Dan R. Littman.
Structural analysis and residue interaction analysis
The interaction modes of the best poses of selected ligands were assessed using the Ligand Interactions module in MOE. The distances between important residues and ligands were measured. Subsequently, the best docking poses of active compounds in complex with CypA were analyzed using IF-E 6.0 [18] (created by Dr. Hooman Shadnia at Carleton University), retrievable from the SVL exchange service. As mentioned in reports by Shadnia and others [18, 19], IF-E 6.0 can partition the native forcefield potentials and derive net interaction forces. It is used to decompose the interaction forces into three dimensions while analyzing the per-residue interactions. A list of positive and negative interaction energy values between a ligand and its neighbor receptor residues (within a defined distance range) can be calculated; negative values indicate favorable interactions, whereas positive values indicate unfavorable interactions. For our candidate ligands, receptor residues oriented less than 4.5 Å were analyzed.
Results and discussion
In silico screening
Comparison between X-ray crystal structure and structure determined by docking
To ensure the validity of our docking procedures and docking parameters before screening, we performed a test using the co-crystallographic data (pdb identifier: 2CYH) of CypA and AP (Fig. 1) [12]. AP was isolated from the complex and then re-docked into the receptor CypA using the MOE software package. The best docking pose to emerge from our docking differed from the original conformation only by 0.261 Å in root mean square deviation (RSMD). Therefore, we selected the pose displaying the lowest docking score as the best pose. Figure 2 shows the comparison between the X-ray crystal structure (colored in red) of AP and the best docking pose (colored in blue).

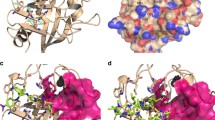
The structure of receptor cyclophilin A (CypA) and its binding sites. a Representation of the dipeptide alanine-proline (AP, red) inside the CypA active site cavity. CypA adopts an eight-stranded antiparallel beta barrel structure, and AP is buried deeply in one of the cavities on the surface of CypA. The structure was obtained from 2CYH.pdb, and the figure was created by Molecular Operating Environment (MOE; Chemical Computing Group, Montreal, Canada) software. b Three docking sites on the surface of CypA. Site A is also known as the CypA MVA11-binding pocket, and AP also binds here. Site B is a hydrophobic pocket that is also known as the CypA Abu2-binding pocket. Above these two sites, there is some space, which we call Site C in this paper

Comparison of the re-docked conformation of AP with the crystallographic conformation in the CypA/AP complex. Blue Re-docked conformation, red crystallographic conformation. The root mean squared deviation (RMSD) between the two conformations was as low as 0.261 Å. The surface of the receptor is colored according to lipophilicity. Green Hydrophobic areas, pink hydrophilic areas
Basic properties of compounds in the target database
We selected a commercially available database from ChemGenesis (http://www.all-chemy.com/) containing 1,377 low-molecular-weight compounds as the source database. The drug-likeness of these compounds was calculated by the QuaSAR-Descriptor module in MOE. More than 85 % of the compounds obeyed drug-likeness (the number of violations of Lipinski’s Rule of Five [13] was less than two). Although less drug-like compounds are usually removed from the target database before docking, because of the small number of compounds in the database, both drug-like and less drug-like compounds were tested. The calculated molecular weights of compounds varied between 201.2 and 622.8 Da, and 1,325 compounds had molecular weights of less than 500 Da. According to the pocket size of CypA, the diameters of the compounds, which varied between 7 and 30 Å, were considered suitable, and no compounds were rejected before performing docking studies.
Analysis of receptor properties and definition of docking sites
According to the co-crystal data of CypA/AP, CypA adopts an eight-stranded antiparallel beta barrel structure (Fig. 1a), and AP is buried deeply in one of the cavities on the surface of CypA (Fig. 1b). This cavity is also where CsA, the HIV CA fragment, and other substrates bind. The most important residues of this cavity were identified by site-directed mutagenesis (Arg55, Phe60, Phe113, and His126) [20]. This cavity has also been called Site A or the MVA11-binding pocket (according to CsA binding mode) in previous reports [8, 21]. Over a saddle-like region, there is another hydrophobic pocket called Site B, or the Abu2-binding pocket. In addition to Sites A and B, there is also some empty space above, referred to here as Site C (Fig. 1b). Although the co-crystallographic data of the CypA/CA fragment indicate that Site A should be the direct docking site, Sites B or C could be considered an auxiliary cavity when compounds possess important elongated functional groups. Therefore, using the SiteFinder module in MOE, we defined a larger docking region contained among Sites A, B and C, and docked compounds in the target database into CypA.
Selection of compounds for biological evaluation
According to the predicted fitting score, which expresses the lowest binding energy of a certain pose, we selected 29 compounds, as shown in Figs. 3, and two previously reported active compounds as controls [8, 22]. We observed two binding modes: (1) molecules covering both Site A and Site B; and (2) molecules placed into Site B and Site C, but not Site A (Fig. 4). Of the 29 compounds listed in Fig. 3, 20 bound in the style of Mode 1 (Fig. 4a), and 9 compounds in the style of Mode 2 (Fig. 4b), as the lowest scored poses.

Control compounds D4 and FD8 and 29 selected compounds docked into the surface sites of CypA. a Mode 1: lowest scored pose of each compound covered the region of Site A and Site B. b Mode 2: lowest scored pose of each compound docked into Site B and Site C, but not Site A


Structures of compounds selected from the docking studies and the control compounds (D4 and FD8). The identifiers of compounds docking in Mode 1 poses are in black, and those of compounds docking in Mode 2 poses are in blue
Site A was confirmed to be responsible for the PPIase activity of CypA and to serve as the binding site of the CA fragment of HIV and CsA, and thus several groups have made efforts to locate compounds that can fill this pocket as much as possible by screening and/or molecular design experiments [8, 21, 22]. Therefore, molecules that interact with CypA in Mode 1 were more likely to be investigated; however, the inhibitory activities of most of the candidates were limited to the micromolar level. To find potent inhibitor candidates with novel skeletons, we kept both Mode 1 and Mode 2 compounds for further biological evaluation.
Experimental results of viral replication and cell viability
Biological assays were performed to determine whether the 29 compounds and the 2 positive controls exerted inhibitory effects on viral replication and induced cellular toxicity. 293T cells were employed as virus-producing cells, whereas a T cell line, MT-4, and a reporter cell line expressing HIV-1 receptors, U87.CD4.CXCR4, were used as viral target cells. This assay system can evaluate the inhibitory effects of test compounds in a single replication cycle of HIV-1. The results demonstrated that most of the other tested compounds either did not exert noticeable inhibitory effects on HIV-1 replication or exhibited strong cytotoxicity at higher concentrations (Table 1). Moreover, although compound 4 exerted an inhibitory effect on viral replication, it displayed low solubility (Table 1); therefore, we did not evaluate this compound further. By contrast, two of our compounds, 12 and 23, as well as the two positive control compounds, exerted potent inhibitory effects on HIV-1 replication while exhibiting low cytotoxicity at the effective concentration inhibiting viral growth by 50 % (Fig. 5). Compared with the control compounds D4 and FD8, our hit compounds exhibited stronger inhibitory activities.

Inhibitory effects on human immunodeficiency virus type 1 (HIV-1) replication and cellular toxicities of active compounds and controls. A vesicular stomatitis virus G protein-pseudotyped reporter virus was produced from 293T cells transfected with proviral DNA in the presence of various concentrations of the compound indicated. U87.CD4.CXCR4 or MT-4 cells were treated with a corresponding compound and then infected with the virus produced in the culture supernatant of 293T cells. Twenty-four hours after infection, the luciferase activity of infected cells was measured. The results are expressed as a percentage of viral replication, which was calculated by determining the reduction in luciferase activity in the presence of a compound compared with that in the control experiment in the absence of the compound. All data points represent the means of two to four independent experiments. In addition, the cytotoxicity of each compound was measured using WST-1 reagent. The cytotoxicity of each compound is expressed as a percentage of the reduction in the absorbance in the presence of the compound
Structural analysis of active compounds
The docking poses of compounds 23 and 12 were different. The best docking pose of 23 utilized Mode 1 binding (Fig. 6a), whereas that of 12 utilized Mode 2 binding (Fig. 6b). The interactions were first assessed using the Ligand Interactions module in MOE. These analyses provided direct schematic views of the interactions of 23 and 12 with CypA, as shown in Fig. 7a and c, respectively. Compound 23 inserted into both Site A and Site B. Figure 7a and b illustrate the dimensional interaction maps and docking poses, respectively, of 23 with CypA. The presence of 2-nitrobenzenesulfonamide appears responsible for the binding between 23 and Site A of CypA. The oxygen atom in the sulfonamide group interacts with the nitrogen atom at the ω position of Arg55 (23: O – CypA: Arg55: Nω) within a distance of 2.9 Å. The aromatic ring of nitrobenzene interacts with both Gln63 and Phe113. Two edge-to-end (T-shaped) hydrogen-aromatic ring interactions (23:aromatic ring–CypA:Gln63:H and 23:aromatic hydrogen–CypA:Phe113:aromatic ring) form a π–π stacking network. Because Arg55 and Phe113 are important pocket-forming residues and are highly conserved in most CypA isolates, these interactions are very meaningful for inhibitor design. Another key interaction revealed in the interaction map is the hydrophobic interaction between Ala103 and the triazole group of 23. For 12, arene-cation interactions (12: benzene ring–CypA: Arg55: NH1 and NH2) were detected. As mentioned above, Arg55 is a critical residue for ligand binding. These interactions can partly explain the activity of 12.

Docking poses of two active compounds: 23 and 12. 23 binds using Mode 1 (a), and 12 binds using Mode 2 (b)

Schematic view and docking pose of compound 23 (a, b) and compound 12 (c, d) with their binding sites. For docking poses, only important residues are displayed
Geometry-based interaction fingerprints provided loosely approximated insights into the fitting of our two active compounds into the receptor sites. However, it is difficult to determine the detailed intermolecular energies, particularly in terms of hydrophobic interactions. Therefore, we performed additional post-docking analyses. The interactions between binding site residues and active compounds were scrutinized using IF-E 6.0 [18] (created by Dr. Hooman Shadnia at Carleton University, http://www.shadnia.com/H_IFE/index.htm), retrievable from the SVL exchange service. This program can differentiate favorable ligand–residue interactions from unfavorable ones by force vectors and energy values. A negative sign indicates a stable interaction, whereas a positive sign indicates an unstable or repulsive interaction. The results of 23 and 12 for each residue near ligand atoms (less than 4.5 Å in distance) are presented in Fig. 8. For 23, in addition to the important residues mentioned above, we can see that Ala101 is also responsible for binding (Fig. 8a). The carbon in the Ala101 backbone is in approximation with the carbonyl oxygen of 23 (separated by 3.57 Å) (Fig. 7a). The dispersion force between the electron-poor environment of the Ala101 backbone carbon and the electron-rich environment of the carbonyl oxygen of 23 resulted in good binding effects. For 12, the negative-signed interaction energy values, which indicate favorable residues for ligand binding, could be detected at multiple residues of CypA including the aforementioned residue Arg55. In addition to Arg55, other favorable residues for binding are listed in Fig. 8b. Conversely, a strong signal for a positive-signed peak could be detected at Ala103, suggesting that it is an unfavorable residue for the binding of 12. By assessing atomic distances, we found that the methyl hydrogen atom of the Ala103 side chain and the aromatic hydrogen atom of 12 are approximately 1.8 Å apart (Fig. 7d). It can be concluded that the two hydrogen atoms clash with each other. However, it should be underlined that this docking pose was obtained from a rigid receptor docking procedure; therefore, this model cannot explain any “induced-fitting” phenomena. We assume that, due to the strong repulsion detected, the receptor residue should shift into a more appropriate orientation in a real system.

Ligand–receptor interaction energy values (per-residue values) calculated using IF-E 6.0. The residue interaction energy data values are expressed in kcal mol−1. a Graph for 23, b graph for 12
Compounds 23 and 12 have similar skeletons; however, they exhibited different docking modes. Molecules that target the combination region of the two modes can be assumed to be more active. These types of combined molecules are now under investigation using in-silico-based methods.
Conclusions
The present work involved the discovery of HIV-1 inhibitors targeting CypA via in silico and biological screening methods. Twenty-nine compounds selected from a database, together with control compounds, were examined for antiviral activities. Two of the compounds exhibited comparatively good effects in biological assays. In particular, compounds 12 and 23 both exhibited anti-HIV-1 activities with relatively low cytotoxicity at the effective concentration inhibiting viral growth by 50 %. From our experimental results, 12 and 23 may be used as lead compounds for novel type of anti-HIV inhibitors, although biochemical experiments to confirm that the target is really CypA are still needed.
References
Ke H, Huai Q (2003) Structures of calcineurin and its complexes with immunophilins-immunosuppressants. Biochem Biophys Res Commun 311:1095–1102
Foster TL, Gallay P, Stonehouse NJ, Harris M (2011) Cyclophilin A interacts with domain II of hepatitis C virus NS5A and stimulates RNA binding in an isomerase-dependent manner. J Virol 85:7460–7464
Chatterji U, Bobardt MD, Lim P, Gallay PA (2010) Cyclophilin A-independent recruitment of NS5A and NS5B into hepatitis C virus replication complexes. J Gen Virol 91:1189–1193
Chen Z, Mi L, Xu J, Yu J, Wang X, Jiang J, Xing J, Shang P, Qian A, Li Y, Shaw PX, Wang J, Duan S, Ding J, Fan C, Zhang Y, Yang Y, Yu X, Feng Q, Li B, Yao X, Zhang Z, Li L, Xue X, Zhu P (2005) Function of HAb18G/CD147 in invasion of host cells by severe acute respiratory syndrome coronavirus. J Infect Dis 191:755–760
Lin TY, Emerman M (2006) Cyclophilin A interacts with diverse lentiviral capsids. Retrovirology 3:70
Zenke G, Strittmatter U, Fuchs S, Quesniaux VF, Brinkmann V, Schuler W, Zurini M, Enz A, Billich A, Sanglier JJ, Fehr T (2001) Sanglifehrin A, a novel cyclophilin-binding compound showing immunosuppressive activity with a new mechanism of action. J Immunol 166:7165–7171
Li Q, Moutiez M, Charbonnier JB, Vaudry K, Menez A, Quemeneur E, Dugave C (2000) Design of a Gag pentapeptide analogue that binds human cyclophilin A more efficiently than the entire capsid protein: new insights for the development of novel anti-HIV-1 drugs. J Med Chem 43:1770–1779
Li J, Tan Z, Tang S, Hewlett I, Pang R, He M, He S, Tian B, Chen K, Yang M (2009) Discovery of dual inhibitors targeting both HIV-1 capsid and human cyclophilin A to inhibit the assembly and uncoating of the viral capsid. Bioorg Med Chem 17:3177–3188
Chen K, Tan Z, He M, Li J, Tang S, Hewlett I, Yu F, Jin Y, Yang M (2010) Structure-activity relationships (SAR) research of thiourea derivatives as dual inhibitors targeting both HIV-1 capsid and human cyclophilin A. Chem Biol Drug Des 76:25–33
Vajdos FF, Yoo S, Houseweart M, Sundquist WI, Hill CP (1997) Crystal structure of cyclophilin A complexed with a binding site peptide from the HIV-1 capsid protein. Protein Sci 6:2297–2307
Mikol V, Kallen J, Pflugl G, Walkinshaw MD (1993) X-ray structure of a monomeric cyclophilin A-cyclosporin A crystal complex at 2.1 A resolution. J Mol Biol 234:1119–1130
Zhao Y, Ke H (1996) Mechanistic implication of crystal structures of the cyclophilin-dipeptide complexes. Biochemistry 35:7362–7368
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23:3–25
Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA (1986) Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291
Tokunaga K, Greenberg ML, Morse MA, Cunning RI, Lyerly HK, Cullen BR (2001) Molecular basis for cell tropism of CXCR4-dependent human immunodeficiency virus type 1 isolates. J Virol 75:6776–6785
Fouchier RA, Meyer BE, Simon JH, Fischer U, Malim MH (1997) HIV-1 infection of non-dividing cells: evidence that the amino-terminal basic region of the viral matrix protein is important for Gag processing but not for post-entry nuclear import. EMBO J 16:4531–4539
Björndal A, Deng H, Jansson M, Fiore JR, Colognesi C, Karlsson A, Albert J, Scarlatti G, Littman DR, Fenyö EM (1997) Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J Virol 71:7478–7487
Shadnia H, Wright JS, Anderson JM (2009) Interaction force diagrams: new insight into ligand-receptor binding. J Comput Aided Mol Des 23:185–194
Dal Ben D, Buccioni M, Lambertucci C, Marucci G, Thomas A, Volpini R, Cristalli G (2010) Molecular modeling study on potent and selective adenosine A(3) receptor agonists. Bioorg Med Chem 18:7923–7930
Zydowsky LD, Etzkorn FA, Chang HY, Ferguson SB, Stolz LA, Ho SI, Walsh CT (1992) Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Protein Sci 1:1092–1099
Guichou JF, Viaud J, Mettling C, Subra G, Lin YL, Chavanieu A (2006) Structure-based design, synthesis, and biological evaluation of novel inhibitors of human cyclophilin A. J Med Chem 49:900–910
Chen S, Zhao X, Tan J, Lu H, Qi Z, Huang Q, Zeng X, Zhang M, Jiang S, Jiang H, Yu L (2007) Structure-based identification of small molecule compounds targeting cell cyclophilin A with anti-HIV-1 activity. Eur J Pharmacol 565:54–59
Acknowledgments
This study was supported by a Health Labor Sciences Research Grant, the Japanese Ministry of Health, Labor and Welfare, the program of the Japan Initiative for Global Research Network on Infectious Diseases (J-GRID) by the Ministry of Education, Cultures, Sports, Science and Technology (MEXT) of Japan, and Grants-in-Aid for the Systematic Graduate School Education Innovation Program (development of health and environment risk management experts) from MEXT of Japan.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Yu-Shi Tian and Chris Verathamjamras contributed equally to this work.
Rights and permissions
About this article
Cite this article
Tian, YS., Verathamjamras, C., Kawashita, N. et al. Discovery of novel low-molecular-weight HIV-1 inhibitors interacting with cyclophilin A using in silico screening and biological evaluations. J Mol Model 19, 465–475 (2013). https://doi.org/10.1007/s00894-012-1560-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-012-1560-7