
Abstract
A comprehensive ab initio investigation using coupled cluster theory with the aug-cc-pVnZ, n = D,T basis sets is carried out to identify distinct structures of the Al4H —14 cluster anion and to evaluate its fragmentation stability. Both thermodynamic and mechanistic aspects of the fragmentation reactions are studied. The observation of this so far the most hydrogenated aluminum tetramer was reported in the recent mass spectrometry study of Li et al. (2010) J Chem Phys 132:241103–241104. The four Al4H —14 anion structures found are chain-like with the multiple-coordinate Al center and can be viewed approximately as comprising Al2H —7 and Al2H7 moieties. Locating computationally some of the Al4H —14 minima on the correlated ab initio potential energy surfaces required the triple-zeta quality basis set to describe adequately the Al multi-coordinate bonding. For the two most stable Al4H —14 isomers, the mechanism of their low-barrier interconversion is described. The dissociation of Al4H —14 into the Al2H —7 and Al2H7 units is predicted to require 20-22 (10-13) kcal mol-1 in terms of ΔH (ΔG) estimated at T = 298.15 K and p = 1 atm. However, Al4H —14 is found to be a metastable species in the gas phase: the H2 loss from the radical moiety of its most favorable isomer is exothermic by 18 kcal mol-1 in terms of ΔH (298.15 K) and by 25 kcal mol-1 in terms of ΔG(298.15 K), with the enthalpic/free energy barrier involved being less than 1 kcal mol-1. By contrast with alane Al4H —14 , only a weakly bound complex between Ga4H —12 and H2 has been identified for the gallium analogue using the relativistic effective core potential.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aluminum-hydrides, or alanes, have been proposed to be potential hydrogen storage media, building blocks for new cluster assemblies and high-energy-density materials [1–6]. During the last three years, several novel alanes, both neutral and anionic, have been made and characterized. The emerging structural and electronic properties of alanes and their derivatives involving a small Al4 cluster core have been notably the focus of the widespread interest [1, 2, 5, 6].
The new hydrogenated Al4H6 neutral cluster was reported to be a stable species in the gas phase based on the results of the photoelectron study of the corresponding anion Al4H —6 and density functional theory (DFT) electronic structure calculations for the neutral [1]. Furthermore, the Al4H6 cluster was suggested to be a high-energy density molecule, with the estimated heat of combustion (giving the Al2O3 and water products) to be about 2.5 times greater than that for methane [1]. Various Al4R6 and Al4R5X derivatives of Al4H6 with the hydrogen atoms substituted by the bulky R groups and halogens X (R = PtBu2; X = Br,Cl) were subsequently prepared and structurally characterized [5].
The whole series of novel aluminum-hydride cluster anions Al4H —n were generated using the laser-induced plasma technique and characterized by mass spectrometry [2]. The most hydrogenated anionic aluminum tetramer reported in Ref. [2] was Al4H —13 . In the related study published recently, the new gas phase AlnH —m clusters were produced with a pulsed arc discharge source and identified using mass spectrometry [6]. The latter investigation reported the aluminum hydride clusters having an extraordinary hydrogen content, with the hydrogen atom number (m)-aluminum atom number (n) ratio exceeding 3: for n = 5-8, the clusters with m = 3n + 1 hydrogen atoms were detected. Most notably, for n = 4, the cluster having m = 3n + 2 hydrogens, or Al4H —14 , was observed according to the authors [6], although the actual structure of this cluster anion with an open electronic shell was not reported.
Previously, using density functional and correlated ab initio methods we compared the structures, thermodynamic stabilities and bonding of the multiply hydrogenated aluminum and gallium neutral clusters M3H9 and M4H12 (M = Al,Ga) [7]. The distinct difference found between the corresponding clusters with M = Al and Ga was the stability of the open type structures called by us “hypervalent”, involving the five- or six-coordinate M atom (in terms of the M-H bonds). We showed specifically that, for M = Al, such a structure was relatively stable, lying within 2 kcal mol-1 of the cyclic global minimum, whereas for M = Ga, the “hypervalent” isomer appeared to be destabilized relative to the lowest energy cyclic species, especially for M4H12 [7].
Herein we present extensive high-level ab initio calculations of the structures and fragmentation stability of Al4H —14 as well as of the reaction paths for interconversion and decomposition of the anion. Notably, for this novel species, we have carefully studied the dependence of the calculated results on the level of theory used. As described below, we have identified four different chain-like structures of Al4H —14 which involve the multiple-coordinate Al center, reminiscent of the structural motif seen in the “hypervalent” isomer of the Al4H12 cluster [7]. In order to assist in the future assignment of the actual isomer of the Al4H —14 anion present in the experimental mass spectrum [6], we have calculated vertical electron detachment energies (VDEs) for the distinct structures. We also show that by contrast to the aluminum hydride Al4H —14 , only a weakly bound complex between the Ga4H —12 anion and H2 has been identified for the gallium counterpart Ga4H —14 .
Computational methods
Coupled cluster theory with single and double excitations (CCSD) [8] and second-order Møller-Plesset perturbation theory (MP2) [9] were used in conjunction with the augmented correlation consistent aug-cc-pVnZ, n = D,T basis sets [10, 11]. All the geometries were optimized and characterized as minima or relevant transition states at each computational level. The MP2 vibrational frequencies were calculated using the analytical Hessians, whereas the CCSD frequencies were computed with the analytical gradients and numerical Hessians. For comparison, optimizations and frequency calculations were also carried out using the same basis sets with the B3LYP density functional [12, 13]. Spin-unrestricted (U) version of each method was used for the open-shell species. To establish accurate energetics, single-point energy calculations were performed at the MP2/aug-cc-pVTZ geometries using CCSD with perturbatively included triples (CCSD(T)) [8] with the aug-cc-pVTZ basis set [10, 11]. In addition, the structures and stability of Al4H —14 were studied using the multi-level G4 scheme [14]. Enthalpies and Gibbs free energies were calculated at T = 298.15 K and p = 1 atm. For the gallium analogue, Ga4H —14 , the energy-consistent effective core potential (ECP), ECP10MDF [15], which replaces the 1s22s22p6 core of Ga by the pseudopotential was used (in addition to the all-electron calculations) to take into account relativistic effects of this atom. The explicitly treated Ga electrons (3s23p63d104s24p1) were described by the associated (11s12p10d2f)/[6s6p5d2f] basis set [16], augmented by diffuse functions (aug-cc-pVTZ-PP). The latter basis was employed in conjunction with the all-electron aug-cc-pVTZ basis sets for H [10, 11, 17]. The relevant computational methods using the ECP10MDF will be referred to as MP2/ECP and B3LYP/ECP. Calculations in the current work were performed employing Gaussian 09 program [18].
Results and discussion
Al4H —14 structures - method dependence
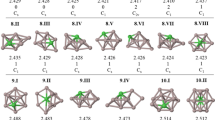
Figure 1 illustrates the optimization level dependence of the four distinct structures of the Al4H —14 cluster anion, S1(2A), S2(2A), S3(2A′) and S3′(2A). These doublet structures are chain-like with the bridging hydrogens. Attempts to locate a cyclic Al4H —14 isomer were unsuccessful. Computations show that S1 exists on the potential energy surfaces calculated at the five levels (with all positive force constants predicted at each level). By contrast, the identification of S2 appeared to be method dependent: this structure has been located on the UB3LYP/aug-cc-pVDZ, UB3LYP/aug-cc-pVTZ and UMP2/aug-cc-pVTZ potential energy surfaces, but not on the UMP2/aug-cc-pVDZ and UCCSD/aug-cc-pVDZ surfaces. In turn, S3 and S3′ structures are potential minima at all the levels, but UMP2/aug-cc-pVDZ. These results indicate that with the correlated methods, the triple-zeta flexibility basis set is required to describe adequately the bonding situation for Al4H —14 .

S1, S2, S3 and S3′ structures of the Al4H —14 anion (distances in Ångstroms) calculated with the correlated ab initio and density functional methods using the aug-cc-pVnZ, n = D,T basis sets. Note that S2 minimum has not been located on the UCCSD/aug-cc-pVDZ and UMP2/aug-cc-pVDZ potential energy surfaces, whereas S3(S3′) minima have not been identified on the UMP2/aug-cc-pVDZ energy surface (see the text)
As seen in Fig. 1, the unique features of the Al4H —14 species are the involvement of the multi-coordinate (Al1 or Al2) atom, the presence of not symmetrical Al-H-Al bridges with the H atoms not shared equally with the Al atoms as well as the presence of the significantly elongated terminal Al1(Al2)-H bonds. It is relevant to note here that multi-coordinate Al centers (participating in the six Al-H-Al bridges) were first observed in the solid state structure of AlH3 polymer in 1969 [19].
Figure 1 also shows that the use of the correlated methods, especially in conjunction with the aug-cc-pVTZ basis set, causes a significant shortening of most of the Al-Al distances (up to 0.3 Å) along with a major decrease of the Al2-H bridging bond of S1 (relative to the UB3LYP results). As the energy calculations indicate (see Table 1), the four Al4H —14 species are of similar stability, lying within 2.2 and 2.4 kcal mol-1 at the most comprehensive UCCSD(T)/aug-cc-pVTZ//UMP2/aug-cc-pVTZ and G4 (0 K) levels, respectively, with S3 being the most stable of the four, followed by S3′.
We suggest that the simplest description of the Al4H —14 anion structure would involve the ‘Al2H —7 ‘ and ‘Al2H7’ units. To support this model we report in Fig. 2 the optimized geometries of the isolated Al2H —7 anion and Al2H7 radical (both structures in Fig. 2 show positive force constants at each computational level). We find further support from the Mulliken spin populations, which indicate that the ‘Al2H7’ fragment of S1 and S3(S3′) is a sole carrier of the spin density. For S2, although its neutral fragment differs most from the isolated Al2H7 geometry (the former matches rather the higher energy Al2H7 isomer [20]), similar to the cases of S1 and S3(S3′), this moiety holds the spin density.

Structures of the isolated Al2H —7 anion and Al2H7 neutral (distances in Ångstroms) calculated with the correlated ab initio and density functional methods using the aug-cc-pVnZ, n = D,T basis sets. Note that Al2H7 minimum has not been located on the UMP2/aug-cc-pVDZ potential energy surface
The results in Fig. 2 further show that an appreciable bending occurs for isolated Al2H —7 at the correlated levels compared to the UB3LYP results. Earlier computational studies of Al2H —7 established this anion to be thermodynamically stable with respect to both the AlH3 + AlH —4 [21] and Al2H6 + H— [22, 23] fragmentations. For isolated Al2H7(2A), we predicted the lowest energy structure to be double-bridged, in agreement with the recent density functional work [20] (note that with UB3LYP/aug-cc-pVTZ, Al2H7 has higher Cs (2A") symmetry). This radical species also features two lengthened terminal Al-H bonds, to 1.66 and 1.76 Å, with a relatively short Al-Al distance of 2.62 Å (the UMP2/aug-cc-pVTZ results), and it bears resemblance with the radical moiety of Al4H —14 , especially for S1. The adiabatic electron detachment energy of the Al2H —7 anion, or the adiabatic electron affinity of the Al2H7 neutral was computed to be relatively high at 4.14 (4.20) eV with the UCCSD(T)/aug-cc-pVTZ//UMP2/aug-cc-pVTZ (G4(0 K)) calculations, pointing to the appreciable electronic stability of the anion.
Interconversion of Al4H —14 isomers
We found that the lowest-energy isomers of Al4H —14 , S3 and S3′, are connected by the transition state S3-S3′ TS, shown in Fig. 3. During this interconversion, Ha and Hb of S3 move in the opposite directions, breaking Cs symmetry (see Fig. 4). Past the S3-S3′ TS, Hb keeps moving toward Hc, whereas Ha moves away from Al2. In the resulting S3′ structure of very similar stability to S3 (Table 1), the new longest terminal Al1-H links are those involving Hb and Hc of 1.67 and 1.87 Å (the UMP2/aug-cc-pVTZ results), with Ha forming “normal” terminal bond, similar to Hd. The S3 → S3-S3′ TS → S3′ reaction path is confirmed by the intrinsic reaction coordinate (IRC) computation. At the UCCSD(T)/aug-cc-pVTZ level, only a tiny barrier for the S3 → S3′ rearrangement is predicted of 0.4 kcal mol-1 (note that in the reverse direction, the barrier does not exist at this level, see the upper left panel in Fig. 4).

A structure of the transition state S3-S3′ TS (distances in Ångstroms) calculated with the correlated ab initio and density functional methods using the aug-cc-pVnZ, n = D,T basis sets. S3-S3′ TS is a saddle point for the Al4H —14 (S3) → Al4H —14 (S3′) interconversion (cf. Fig. 4); the associated imaginary frequency is given at each computational level

Illustration of the Al4H —14 (S3) → Al4H —14 (S3′) interconversion occurring through the transition state S3-S3′ TS. The interconversion barrier values shown have been corrected for the zero-point vibrational energies; the value before (after) slash is for the S3→S3′ (S3′→S3) rearrangement
Fragmentation stability of Al4H —14
We next assessed the fragmentation stability of the Al4H —14 anion. The computed energies of the fragmentation reactions of the four isomers of Al4H —14 are reported in Table 2, while the corresponding thermodynamic values are given in Table 3. We have considered three unimolecular decomposition channels: (1) Al4H —14 → Al2H —7 + Al2H7, (2) Al4H —14 → Al4H —13 + H and (3) Al4H —14 → Al4H —12 + H2. The choice of decomposition (1) is justified by the Al4H —14 anion “bonding model” proposed above. The H loss reaction (2) affords the closed-shell anion Al4H —13 , observed in the gas phase by two research groups [2, 6], which is indicative of a relatively stable species. With the fragmentation channel (3) we examine if Al4H —14 is stable with respect to loss of H2 molecule of significant stability.
The last column of Table 2 which compares our most extended zero-point energy corrected UCCSD(T)/aug-cc-pVTZ//UMP2/aug-cc-pVTZ reaction energies with the G4(0 K) values (the latter are given in parentheses) shows a very good agreement between both kinds of results. Therefore, the enthalpies ΔH (298.15 K) and free energies ΔG(298.15 K) presented in Table 3 and discussed in this section were estimated at the G4 level. Regarding decomposition (1), Table 3 indicates that this reaction is always endothermic, by 19.6-22.3 (10.1-13.0) kcal mol-1 with ΔH (298.15 K) (ΔG(298.15 K)). With respect to the H loss reaction (2) and in terms of ΔH (298.15 K), we found it to be slightly endothermic for S3, essentially thermoneutral for S1 and S3′ and insignificantly exothermic for S2. With ΔG(298.15 K), this reaction is moderately exothermic for all the Al4H —14 species, by 4.4-7.3 kcal mol-1. Note that the calculated chain-like structure of the Al4H —13 decomposition product for reaction (2) is reported in the upper panel in Fig. 5. The alternative Al4H13 + H— dissociation process is energetically much less favorable, by about 90 kcal mol-1. This is explained by the considerably smaller electron affinity of H compared to Al4H13, 0.9 vs. 4.8 eV.

Structures of the Al4H —13 and Al4H —12 anions (distances in Ångstroms), being the products of H and H2 loss from Al4H —14 , respectively, calculated with the correlated ab initio and density functional methods using the aug-cc-pVnZ, n = D,T basis sets
By contrast with the two former fragmentations, the reaction of H2 elimination from Al4H —14 (decomposition (3)) is found to be significantly exothermic, by 17.8-20.5 kcal mol-1 in terms of ΔH(298.15 K) and by 24.8-27.7 kcal mol-1 in terms of ΔG(298.15 K) (Table 3). These results indicate that the Al4H —14 anion is thermodynamically disfavored. The structure of the corresponding dehydrogenation product, Al4H —12 , is shown in the lower panel in Fig. 5. In the next section we are reporting the related dehydrogenation pathway.
Reaction pathway for H2 elimination from Al4H —14 (S3)
The results of the previous section showed clearly that H2 elimination from Al4H —14 is by far the most thermodynamically favorable unimolecular decomposition of this anion. To assess kinetic stability of Al4H —14 , we have calculated the reaction pathway for H2 loss occurring from the lowest energy S3 isomer. Due to the proximity of Ha and Hb in Al4H —14 (S3) (cf. Fig. 4), it is anticipated that the H2 elimination will occur relatively easily from its radical moiety. Computations confirm this assumption. The Al4H —14 (S3) → Al4H —12 + H2 reaction has an early and asynchronic transition state, H 2 el. TS (see Fig. 6). At the TS, only Al1-Hb increases moderately relative to that in the reactant, by 0.14-0.16 Å, whereas Al1-Ha actually decreases somewhat, with a hydrogen molecule (Ha-Hb) formed as described by Ha-Hb distance of 1.10-1.18 Å and corresponding imaginary frequency values of 644-751i cm-1.

A structure of the transition state H 2 el. TS for H2 elimination from Al4H —14 (S 3 ) (distances in Ångstroms), calculated with the correlated ab initio and density functional methods using the aug-cc-pVnZ, n = D,T basis sets; the associated imaginary frequency is given at each computational level
Figure 7 summarizes the profiles of ΔH(298.15 K) for the three unimolecular fragmentations of Al4H —14 (S3) considered, with the corresponding ΔG(298.15 K) values given therein in parentheses. The most significant feature of these profiles is that there is only a tiny enthalpic/free energy barrier for H2 loss, being 0.5/0.1 kcal mol-1, suggesting that this elimination is remarkably facile (see also Table 4). This result, combined with the exothermicity of the process predicted for S3 by 18 kcal mol-1 in terms of ΔH(298.15 K) and by 25 kcal mol-1 in terms of ΔG(298.15 K) suggests the Al4H —14 cluster to be best described as a metastable species in the gas phase.

Schematic profiles of ΔH (298.15 K) for the three fragmentation channels of the Al4H —14 (S3) anion (in kcal mol-1); the corresponding ΔG (298.15 K) values are given in parentheses (G4 results). Note that the Al4H —12 …H2 post-reaction complex (shown in Fig. 8) has been omitted in the H2 elimination path
The post-reaction van der Waals complex between Al4H —12 and H2 has also been identified as reported in Fig. 8. This complex, denoted Al4H —12 …H2, was optimized at all five computational levels. Figure 8 shows that both the actual position of the H2 subsystem within the complex and interaction distance(s) are method dependent. With UMP2/aug-cc-pVTZ, describing best long-range dispersion (at the optimization level), the complex has a structure in which the hydride fragment (Al2-H) of Al4H —12 interacts with the slightly polarized H-H bond (based on the Mulliken charges). Furthermore, the UMP2/aug-cc-pVTZ calculated interacting distance of 2.53 Å is distinctly reduced relative to those predicted with the other methods. The UCCSD(T)/aug-cc-pVTZ//UMP2/aug-cc-pVTZ binding energy (De) of Al4H —12 …H2 amounts to 0.7 kcal mol-1. Note that this value coincides with the UMP2/aug-cc-pVTZ result indicating the adequate description of dispersion already at the latter level (for comparison of the six De values of Al4H —12 …H2, see lower panel in Fig. 8).

A structure (distances in Ångstroms) and binding energy (De, kcal mol-1) of the complex Al4H —12 …H2; De has been calculated as [E(Al4H —12 ) + E(H2) – E(Al4H —12 …H2)]
VDE energies
The VDE energies of the four hydride structures of the Al4H —14 anion computed at the UCCSD(T)/aug-cc-pVTZ//UMP2/aug-cc-pVTZ level are compared in Table 5. For the S1, S2, S3 and S3′ species the respective values are 4.83, 5.23, 4.72 and 4.77 eV. These VDEs are found to be even larger than the (adiabatic) electron detachment energy of the Al2H —7 anion discussed above. On the other hand, except for the S2 result, the remaining three VDEs are quite similar and might not help to assist in the future assignment of the actual isomer of the Al4H —14 anion .
Does Ga4H —14 galane exist?
Following the earlier work on comparing the structures, stabilities and bonding of the hydrogenated aluminum and gallium clusters [7], we next studied computationally Ga4H —14 cluster anion. Although at the all-electron DFT UB3LYP/aug-cc-pVTZ level we located the Ga4H —14 minimum analogue of Al4H —14 (S3) (Fig. 9), neither our UB3LYP/ECP optimization nor that using the correlated UMP2/ECP method confirmed this result (the last two approaches employed the relativistic effective core potential ECP10MDF, see the footnote under Fig. 9). Similarly, our attempts to optimize the Ga4H —14 counterparts of S1 and S2 led instead to the van der Waals complex Ga4H —12 …H2, depicted in Fig. 10. As in Al4H —12 …H2, the location of H2 unit within Ga4H —12 …H2 and intermolecular separation are method dependent. Expectedly, the shortest separation between Ga4H —12 and H2 is calculated with UMP2/aug-cc-pVDZ and UMP2/ECP (the highest correlated optimization levels used for the complex). At the former geometry, the UCCSD(T)/aug-cc-pVTZ binding energy (De) of Ga4H —12 …H2 is 1.3 kcal mol-1 (see the lower panel in Fig. 10). As Fig. 10 shows additionally, the use of the relativistic ECP resulted in a significant shortening of the terminal Ga-H bonds compared to the all-electron results (this is actually a joint ECP/basis set effect).

A structure of the Ga4H —14 analogue of Al4H —14 (S3) (distances in Ångstroms) calculated at the UB3LYP/aug-cc-pVTZ and UB3LYP/ECP levels. This structure appeared to be a minimum only with the all electron UB3LYP/aug-cc-pVTZ. No Ga4H —14 minimum has been identified with either UB3LYP/ECP (the first-order saddle point with the imaginary frequency of 40i cm-1 was obtained at this level with the stringent optimization criteria and ‘ultrafine’ grid) or UMP2/ECP (H dissociation occurred from Ga1 during optimization)

A structure (distances in Ångstroms) and binding energy (De, kcal mol-1) of the complex Ga4H —12 …H2 calculated at the all-electron and ECP levels; De has been calculated as [E(Ga4H —12 ) + E(H2) – E(Ga4H —12 …H2)] - note that the UCCSD(T)/aug-cc-pVTZ value of De has been computed at the UMP2/aug-cc-pVDZ optimized structures
The differences noticed here between the Al4H —14 and Ga4H —14 cluster anions are consistent with our recent symmetry-adapted perturbation theory (SAPT) analysis of the aluminum and gallium species pointing out to much stronger “hydride” character of the former [24]. The larger propensity of Al atom for hypercoordinate bonding situations compared to Ga [25] is a relevant qualitative explanation.
Conclusions
For the first time, distinct minima structures of the hydrogen-rich alane Al4H —14 , experimental observation of which was recently reported [6], have been identified at the correlated ab initio levels, and thermodynamic and kinetic stability of this species was assessed at T = 0 K and T = 298.15 K. The structures found are chain-like, contain the multiple-coordinate Al center and approximately comprise the Al2H —7 and Al2H7 moieties. Locating computationally some of the Al4H —14 minima on the UCCSD and UMP2 potential energy surfaces required the triple-zeta quality basis set to describe adequately the Al multi-coordinate bonding. The dissociation of Al4H —14 into the Al2H —7 and Al2H7 units is predicted to require 20-22 (10-13) kcal mol-1 with ΔH(298.15 K) (ΔG(298.15 K)). However, Al4H —14 is predicted to be metastable, because H2 loss from its most favorable S3 isomer is exothermic by 18 kcal mol-1 in terms of ΔH(298.15 K) and by 25 kcal mol-1 in terms of ΔG(298.15 K), with the enthalpic/free energy barrier involved being less than 1 kcal mol-1. The global minimum on the Al4H —14 anion energy surface corresponds to the weakly bound complex Al4H —12 …H2. This kind of complex appeared to be the only minimum structure identified for the gallium counterpart Ga4H —14 , when Ga relativistic effective core potential was used.
In the original experimental study, Li et al. [6] reported that they could observe strong AlnH —m intensities for m > 3n “under some source conditions”. Our current thermodynamic and kinetic (fragmentation reaction barrier) considerations suggest that due to the predicted Al4H —14 metastability in the gas phase, this ion would in fact be detected only under the special experimental conditions.
References
Li X, Grubisic A, Stokes ST, Cordes J, Ganteför GF, Bowen KH, Kiran B, Willis M, Jena P, Burgert R, Schnöckel H (2007) Science 315:356–358
Roach PJ, Reber AC, Woodward WH, Khanna SN, Castleman AW Jr (2007) Proc Natl Acad Sci USA 104:14565–14569
Grubisic A, Li X, Stokes ST, Cordes J, Ganteför GF, Bowen KH, Kiran B, Jena P, Burgert R, Schnöckel HJ (2007) J Am Chem Soc 129:5969–5975
Bazyn T, Krier H, Glumac N, Wang X, Jackson TL (2007) J Prop Power 23:457–464
Henke P, Huber M, Steiner J, Bowen KH, Eichhorn B, Schnöckel H (2009) J Am Chem Soc 131:5698–5704
Li X, Grubisic A, Bowen KH, Kandalam AK, Kiran B, Gantefoer GF, Jena P (2010) J Chem Phys 132:241103–241104
Moc J, Bober K, Mierzwicki K (2006) Chem Phys 327:247–260
Pople JA, Head-Gordon M, Raghavachari K (1987) J Chem Phys 87:5968–5975
Møller C, Plesset MS (1934) Phys Rev 46:618–622
Dunning TH (1989) J Chem Phys 90:1007–1023
Woon DE, Dunning TH (1993) J Chem Phys 98:1358–1371
Becke AD (1993) J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789
Curtiss LA, Redfern PC, Raghavachari K (2007) J Chem Phys 126:084108–084112
Metz B, Stoll H, Dolg M (2000) J Chem Phys 113:2563–2569
Peterson KA (2003) J Chem Phys 119:11099–11112
Kendall RA, Dunning TH, Harrison RJ (1992) J Chem Phys 96:6796–6806
Frisch MJ et al (2010) Gaussian 09, Revision B01. Gaussian Inc, Wallingford, CT
Turley JW, Rinn HW (1969) Inorg Chem 8:18–22
Cui YH, Wang JG, Xu W (2010) Nanotechnology 21:025702
Chiles RA, Dykstra CE (1982) Chem Phys Lett 92:471–473
Charkin OP (2007) Russ J Inorg Chem 52:1925–1936
Goebbert DJ, Hernandez H, Francisco JS, Wenthold PG (2005) J Am Chem Soc 127:11684–11689
Moc J, Bober K, Panek J (2005) J Mol Model 12:93–100
Duke BJ, Liang C, Schaefer HF III (1991) J Am Chem Soc 113:2884–2890
Acknowledgments
The author gratefully acknowledges computational resources provided by the Wroclaw Centre for Networking and Supercomputing, WCSS.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Moc, J. Does Al4H —14 cluster anion exist? High-level ab initio study. J Mol Model 18, 3427–3438 (2012). https://doi.org/10.1007/s00894-012-1353-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-012-1353-z