
Abstract
Structure and energy calculations of pristine and COOH-modified model single wall carbon nanotubes (SWCNTs) of different length were performed at B3LYP/6-31G* level of theory. From 1 to 9 COOH groups were added at the end of the nanotube. The differences in structure and energetics of partially and fully functionalized SWCNTs at one end of the nanotube are observed. Up to nine COOH groups could be added at one end of (9,0) zigzag SWCNT in case of full functionalization. However, for (5,5) armchair SWCNT, the full functionalization was impossible due to steric crowding and rim deformation. The dependence of substituent attachment energy on the number of substituents at the carbon nanotube rim was observed.

Structure and energy calculations of pristine and COOH-modified model single wall carbon nanotubes (SWCNTs) of different length were performed at B3LYP/6-31G* level of theory and from 1 to 9 COOH groups was added at the end of the nanotube
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Three different types of carbon nanotubes are experimentally observed: armchair, zigzag and chiral [1]. These carbon structures are finished with semispheres containing pentagons and hexagons, being formally parts of fullerenes. Due to their structure, the CNTs are hydrophobic, strongly interact with light and possess interesting electrical and physical properties [1–3].
Modification of hydrophobic carbon nanotubes by allowing stronger intermolecular interactions, leading to solubility is expected upon addition of selected small molecules covalently bonded to the (a) end, (b) surface, or (c) both the end and surface (mixed) of SWCNTs [2, 4–13]. The rim structure of SWCNT in case of zigzag or armchair open ended CNT shows a different pattern. Zigzag carbon nanotubes show metallic or semiconductor properties and their ends shows “saw-tooth” like shape. Functionalized carbon nanotubes are promising candidates in material sciences and nanomedicine [1–3]. For example, OH, COOH or NH2 functionalized CNT are easily transformed and could bear longer chains connecting antibodies or drugs. End-substituted SWCNT are by-products of mild oxidation and carboxylic, carbonyl and hydroxyl groups are frequently formed. Most previous works [4–6] concentrated on single functionalized SWCNTs and the impact of substituent on physical properties of modified versus pristine structure (for example, density of states, HOMO-LUMO gap). The pristine CNTs are insoluble in water and organic solvents and this is a serious hindrance in their industrial applications, for example, as efficient nanocomposites [1, 14, 15].
Earlier works demonstrated a possibility of transforming inert and hydrophobic CNTs, into soluble forms [14–17]. This was accomplished by subsequent chemical modification of water soluble CNTs, containing COOH groups [14–17]. Unfortunately, little is known about the systematic changes of energy of zigzag and armchair SWCNTs upon consecutive replacement of rim hydrogen atoms by COOH groups.
Density functional theory (DFT) and, in particular, the exchange-correlation B3LYP hybrid density functional is widely used in molecular modeling studies to predict structure, spectroscopic parameters and energy changes of small, middle and large size molecules [7, 8, 18–20]. Due to the large size of CNTs, the DFT calculations with relatively small basis sets (3-21G or 6-31G*), and also AM1 and PM3 semiempirical methods, have been used for theoretical description of molecular structure and other parameters of finite models of CNTs [8].
In this study, as an extension of our previous works on hydroxyl substituted SWCNTs [19, 21, 22], we would like to get a more detailed information at the atomistic level on the open-end CNT modification with COOH, up to full substitution with nine (zigzag) or ten (armchair) carboxylic groups.
Computational methods
All calculations were performed using Gaussian 09 program [23]. Reliable exchange-correlational B3LYP hybrid density functional and basis sets of relatively small size (3-21G and 6-31G*), enabling completing fairly large scale calculations were selected. Full structure optimization of unsubstituted open-ended (with dangling bonds on carbon saturated with hydrogen atoms), and COOH-modified SWCNTs were performed. Several models of SWCNT were selected, including (9,0) zigzag and (5,5) armchair structures with one and three layers (strings) of hexagon units. IR and Raman harmonic frequencies were calculated in case of one layer with one to 9/10 COOH substituents. All positive frequencies ensured ground state structure of the optimized system.
For comparison purposes only, the calculations with small model molecules including methane, benzene, anthracene and phenanthrene before, and after replacing one hydrogen atom with the carboxyl group were performed at the same level of theory. In addition, to verify the basis set quality impact, these calculations were performed with a large basis set (6-311++G(3df,2pd).
Energy of one COOH group formation at nanotube terminated initially with H atoms was calculated by considering a hypothetical reaction:
Energies of adding subsequent groups (ΔE in kcal mol-1) were assumed as follows:
where n = 1,2,…9 (10).
Initially, the calculations were conducted at B3LYP/3-21G level of theory. Qualitatively, the changes in energies obtained with smaller basis set (3-21G) were similar to those, obtained at B3LYP/6-31G*. Thus, the final results, obtained with the larger basis set will be only discussed.
Results and discussion
The carboxylic group can be considered as an asymmetric substituent with two different ends (O atom vs. OH group) and their position in respect to the rim of the CNT be positioned in a way which minimizes the interactions with the neighboring H-atoms and/or forms H-bonds with other COOH substituents. This was tested on model systems (Scheme 1) by rotating the COOH substituent relative to the aromatic ring plane (changing the dihedral angle Cring–Cring–C = O).

Small model molecules (benzoic, anthracene-9-carboxylic and phenathrene-4-carboxylic acids
The energy landscape of COOH rotation in case of monosubstituted benzene, phenanthrene and anthracene are shown in Fig. 1. In case of benzene, the energy minimum, corresponding to favorable carboxylic orientation, coplanar with the ring, is observed and the perpendicular position, e.g., at both sides of the ring, are about 8 kcal mol-1 higher. In case of anthracene, the energy minimum corresponds to about 45 degree deviation of COOH plane from rings plane and there are also two maxima of the same height (at about 3 kcal mol-1) for the perpendicular orientation. The rotation of COOH in phenanthrene at position 4 (see Scheme 1) leads to an asymmetric shape of energy curve. The basis set effect on the position of energy maxima upon COOH rotation in C6H5COOH is also shown in Fig. 1a. Thus, upon improving the basis set quality from 6-31 G* to 6-311++G** and 6-311++G(3df,2pd) the barrier height slightly decreases (from 7.85 to 6.73 and 6.52 kcal mol-1). It is apparent that the barrier height decreases by about 1.3 kcal mol-1 upon significantly improving the basis set quality.

Energy change upon rotation of COOH substituent relative to ring plane in (a) benzoic acid and (b) anthracene-9-carboxylic acid and phenathrene-4-carboxylic acid
In Fig. 2a and b are shown optimized structures of zigzag and armchair CNT consisting of three ring layers with a single COOH substituent at the rim.

Optimized structures of model (a) zigzag and (b) armchair SWCNTs with a single COOH substituent at the rim (dimensions in Å)
In Fig. 3 are shown energy landscapes of single COOH group rotation attached to zigzag and armchair SWCNTs. In this case, the preferred geometry is observed for both -OH and = O ends of carboxylic group outside the tube (on the circumference). Two energy maxima are observed for COOH group oriented along the tube radius and the slightly lower one corresponds to OH being outside the tube. In case of zigzag CNT, the energy minimum corresponds to C–C–C = O angle of about −5 degrees, (substituent on the circumference) and the highest maximum corresponds to about −90 degrees (C = O outside the tube). The other maximum (with C = O inside, or oriented toward the tube center) is slightly lower. Similarly to Fig. 1a, the improvement of basis set quality from 3-21G to 6-31G* leads to energy barrier lowering by about 4 kcal mol-1. In the case of carboxylic group rotation at the armchair rim, the situation is similar and the corresponding barrier heights are 9 and 7 kcal mol-1 and the energy minimum is observed at about −10 degrees.

Energy change upon rotation of COOH substituent at the rim of model (a) zigzag and (b) armchair SWCNTs
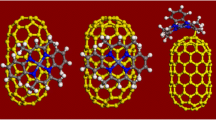
Up to nine carboxylic substituents were placed consecutively at the zigzag rim (see Fig. 4a), forming stable structures. In this case, a kind of threefold symmetry was observed. Nevertheless, some funnel shape deformation and increase of the tube-end diameter was observed.

Optimized structures of model (a) zigzag and (b) armchair SWCNTs fully functionalized with COOH substituents at the rim. Threefold symmetry is indicated for zigzag nanotube
The armchair model consisting of three hexagon layers with up to nine COOH groups at one rim was also stable. In addition, upon complete functionalization of one hexagon layer (the shortest armchair nanotube model) a stable system was also observed (Fig. 4b). However, all attempts to obtain fully functionalized one end of a longer tube, containing three layers of hexagons, failed. This was probably due to steric crowding at the relatively rigid tube skeleton end.
In the next step, starting from model systems of methane and benzene, we examined the energetics of substitution process calculated according to Eq. 2 (see Table 1). Addition of ZPV correction changes the substitution energy slightly while hydroxylation is more favorable in the case of benzene.
The relative carboxylation energy, calculated with Eq. 2 vs. the number of COOH substituents for zigzag and armchair are displaced in Fig. 5a and b.

Dependence of B3LYP predicted carboxylation energy according to Eq. 2 for model zigzag and (b) armchair SWCNTs functionalization at two basis set sizes. For better visualization the data points are connected
It is evident from Fig. 5a that the carboxylation energy for the first hydrogen atom at the rim of zigzag nanotube formed from three layers is about −13 kcal mol-1. This differs from the calculated previously [19] hydroxylation energy of about −35 kcal mol-1. Addition of the second COOH group differs by about 10 kcal mol-1. Significantly smaller energy increments are needed for adding three to nine carboxylic groups.
In Fig. 5b is shown a similar carboxylation energy dependence on replacement of consecutive hydrogen atoms at the rim of armchair CNT model. The first carboxylation energy is higher than for zigzag model (about –8.5 vs. –13 kcal mol-1) whereas for the second group this energy is about −1 vs. –3 kcal mol-1, for armchair and zigzag models, respectively. However, some oscillation of energy is observed, with minima located at odd numbers of COOH. This resembles the results for hydroxylation energy pattern observed previously for armchair CNT [21, 22]. The reason of this behavior was explained earlier as a result of different H-bond ring pattern formation at the rim. The results presented in Fig. 5 indicate higher reactivity of zigzag versus armchair SWCNT rim toward carboxylation. This is in agreement with earlier observations by Kim et al. [24].
Conclusions
The present density theory studies using B3LYP hybrid functional indicate a possibility of COOH-functionalization of one end of zigzag CNT with one to nine substituents. However, the end of armchair nanotube cannot be fully functionalized with COOH groups (one hydrogen atom remains unsubstituted).
On the basis of the performed B3LYP/6-31G* calculations it appears that the replacement of one hydrogen atom at the rim of the zigzag CNT model is a more exothermic process than for armchair model (−13 vs −8.5 kcal mol-1). This indicates a higher reactivity of zigzag CNT toward carboxylation. A gradual and nearly linear increase of energy is observed for subsequent carboxylation, starting from two to nine groups on a zigzag nanotube end.
References
Saito R, Dresselhaus MS, Dresselhaus G (1998) Physical properties of carbon nanotubes. Imperial College Press, London
Khabashesku VN, Margrave JL, Barrera EV (2005) Functionalized carbon nanotubes and nanodiamonds for engineering and biomedical applications. Diamond Rel Mat 14:859–866
Calvert P (1999) A recipe for strength. Nature 399:210–211
Kar T, Akdim B, Duan X, Pachter R (2006) Open-ended modified single-wall carbon nanotubes: A theoretical study of the effects of purification. Chem Phys Lett 423:126–130
Veloso MV, Filho AGS, Filho JM, Fagan SB, Mota R (2006) Ab initio study of covalently functionalyzed carbon nanotubes. Chem Phys Lett 430:71–74
Kuzmany H, Kukovecz A, Simon F, Holzweber M, Kramberger C, Pichler T (2004) Functionalization of carbon nanotubes. Synth Met 114:113–122
Kar T, Scheiner S, Roy AK (2008) The effect on acidity of size and shape of carboxylated single-wall carbon nanotubes. A DFT-SLDB study. Chem Phys Lett 460:225–229
Wongchoosuk C, Udomvech A, Kerdcharoen T (2009) The geometrical and electronic structures of open-end fully functionalized single-walled carbon nanotubes. Curr Appl Phys 9:352–358
Kar T, Scheiner S, Patnaik SS, Bettinger HF, Roy AK (2010) IR characterization of tip-functionalized single-wall carbon nanotubes. J Phys Chem C 114:20955–20961
Kar T, Adkim B, Duan X, Pachter R (2006) Open-ended modified single-wall carbon nanotubes: A theoretical study of the effects of purification. Chem Phys Lett 423:126–130
Salzmann CG, Llewellyn SA, Tobias G, Ward MAH, Huh Y, Green MLH (2007) The role of carboxylated carbonaceous fragments in the functionalization and spectroscopy of a single-walled carbon-nanotube material. Adv Mater 19:883–887
Zhao J, Lu JP, Han J, Yang CK (2003) Noncovalent functionalization of carbon nanotubes by aromatic organic molecules. Appl Phys Lett 82:3746–3751
Vigoloa B, Mamane V, Valsaque F, Le TNH, Thabit J, Ghanbaja J, Aranda L, Fort Y, McRae E (2009) Evidence of sidewall covalent functionalization of single-walled carbon nanotubes and its advantages for composite processing. Carbon 47:411–419
Tasis D, Tagmatarchis N, Georgakilas V, Prato M (2003) Soluble carbon nanotubes. Chem Eur J 9:4000–4008
Sun YP, Fu K, Lin Y, Huang W (2002) Functionalized carbon nanotubes: Properties and applications. Acc Chem Res 35:1096–1104
Chen J, Hamon MA, Hu H, Chen Y, Rao AM, Eklund PC, Haddon RC (1998) Solution properties of single-walled carbon nanotubes. Science 282:95–98
Liu J, Rinzler AG, Dai H, Hafner JH, Bradley RK, Boul PJ, Lu A, Iverson T, Shelimov K, Huffman CB, Rodriguez-Macias F, Shon YS, Lee TR, Colbert DT, Smalley RE (1998) Fullerene pipes. Science 280:1253–1256
Foresman JB, Frisch A (eds) (1996) Exploring chemistry with electronic structure methods. Gaussian Inc, Pittsburg, PA
Chełmecka E, Pasterny K, Kupka T, Stobiński L (2010) Density functional theory studies of OH-modified open-ended single-wall zigzag carbon nanotubes (SWCNTs). J Mol Struct Theochem 948:93–98
Stobinski L, Tomasik P, Lii CY, Chan HH, Lin HM, Liu HL, Kao CT, Lu KS (2003) Single-walled carbon nanotube-amylopectin complexes. Carbohydr Polym 51:311–316
Chełmecka E, Pasterny K, Kupka T, Stobiński L (2011) DFT studies of OH-functionalized open-ended zigzag, armchair, and chiral single wall carbon nanotubes. Phys Status Solidi A 208:1774–1777
Chełmecka E, Pasterny K, Kupka T, Stobiński L (2011) OH-functionalized open-ended single wall armchair carbon nanotubes (SWCNT) studied by density functional theory. J Mol Model. doi:10.1007/s00894-011-1181-6
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin R. L, Fox DJ (2009) Gaussian 09, Revision A.02. Gaussian. Wallingford, CT
Kim C, Seo K, Kim B, Park N, Choi YS, Park KA, Lee YH (2003) Tip-functionalized carbon nanotubes under electric fields. Phys Rev B 68:115403(1–7)
Acknowledgments
This work was supported by the Polish Council for Science through the Development Grants for the years 2008–2011 (NR 15-0011-04/2008, KB/72/13447/IT1-B/U/08) and the calculations were performed using a hardware and software at WCSS Wrocław. We also acknowledge the kind support of Medical University of Silesia, University of Silesia and University of Opole and fruitful discussions with Prof. Małgorzata Broda.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Chełmecka, E., Pasterny, K., Kupka, T. et al. DFT studies of COOH tip-functionalized zigzag and armchair single wall carbon nanotubes. J Mol Model 18, 2241–2246 (2012). https://doi.org/10.1007/s00894-011-1242-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-011-1242-x