
Abstract
The serotonin (5-HT) transporter (SERT) plays an important role in the termination of 5-HT-mediated neurotransmission by transporting 5-HT away from the synaptic cleft and into the presynaptic neuron. In addition, SERT is the main target for antidepressant drugs, including the selective serotonin reuptake inhibitors (SSRIs). The three-dimensional (3D) structure of SERT has not yet been determined, and little is known about the molecular mechanisms of substrate binding and transport, though such information is very important for the development of new antidepressant drugs. In this study, a homology model of SERT was constructed based on the 3D structure of a prokaryotic homologous leucine transporter (LeuT) (PDB id: 2A65). Eleven tryptamine derivates (including 5-HT) and the SSRI (S)-citalopram were docked into the putative substrate binding site, and two possible binding modes of the ligands were found. To study the conformational effect that ligand binding may have on SERT, two SERT–5-HT and two SERT–(S)-citalopram complexes, as well as the SERT apo structure, were embedded in POPC lipid bilayers and comparative molecular dynamics (MD) simulations were performed. Our results show that 5-HT in the SERT–5-HTB complex induced larger conformational changes in the cytoplasmic parts of the transmembrane helices of SERT than any of the other ligands. Based on these results, we suggest that the formation and breakage of ionic interactions with amino acids in transmembrane helices 6 and 8 and intracellular loop 1 may be of importance for substrate translocation.

SERT–5-HTB binding mode
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The serotonin [5-hydroxytryptamine (5-HT)] transporter (SERT) is located in the membrane of presynaptic neurons and plays an important role in the termination of serotonergic neurotransmission by transporting 5-HT from the synaptic cleft into the presynaptic neuron. SERT, and the closely related dopamine and noradrenaline (norepinephrine) transporters (DAT and NET, respectively), are located in limbic areas of the CNS that are involved in mood, emotion and reward processes, and are important targets of therapeutic drugs as well as psychoactive illicit drugs. Among the compounds that act on SERT are drugs belonging to the two main groups of antidepressants—the classic tricyclic antidepressants (TCAs) and the newer selective serotonin reuptake inhibitors (SSRIs)—and well-known drugs of abuse such as cocaine and amphetamines, including 3,4-methylenedioxy-N-methamphetamine (MDMA, commonly known as “ecstasy”).
SERT, DAT and NET belong to the neurotransmitter/sodium symporter (NSS) transporter family (Transporter Classification code 2.A.22 [1]), also known as the SLC6 family [2]. This transporter family constitutes a large number of secondary transporters that use Na+ electrochemical gradients to transport extracellular solutes across membranes. At least 177 eukaryotic and 167 prokaryotic transporters have been classified as belonging to this family [3], transporting a large number of solutes. In addition to the biogenic amines, amino acids such as γ-aminobutyric acid (GABA), glycine, tryptophan, tyrosine and leucine (the GAT-1, GlyT, TnT, Tyt1 and LeuT transporters, respectively) are transported by NSS transporters [1].
The three-dimensional (3D) structure of SERT (or, indeed, that of any eukaryotic NSS family member) has not been experimentally determined; however, the first X-ray crystal structure of a prokaryotic NSS family member, the Aquifex aeolicus leucine transporter (LeuT), was published in 2005 [4]. Since then, several crystal structures of LeuT have been published, and 3D structures of LeuT in an occluded conformation [5–7] and in an outward-facing conformation [8] are now available. These crystal structures can be used as templates for the generation of 3D models of SERT and other NSS transporters using the homology modeling approach, taking advantage of the fact that 3D structure is more conserved than the sequence [9]. Several SERT models have been generated based on the occluded LeuT crystal structure [10–12] and a published comprehensive alignment of NSS family members by Beuming et al. [3].
In 1966, transporter proteins were suggested to operate through an alternating-access mechanism [13] in which a central substrate binding site is alternately exposed to either the extracellular environment or the cytoplasm through conformational changes of the protein. The 3D crystal structures of LeuT thus fit this proposed transport mechanism, as they are in open-to-out and occluded conformations [4–8]. In the latter conformation, leucine is bound in the substrate binding site of LeuT, and the side chains of two phenylalanine residues (corresponding to Y176 and F335 in SERT) and one arginine and glutamate residue (corresponding to R104 and E493 in SERT) block access from the extracellular environment to the substrate binding site [4–7]. In the outward-facing conformation, the competitive inhibitor L-tryptophan displaces leucine from the substrate binding site and causes LeuT to stabilize in an outward-facing conformation, where the distance between the side chains of Y176 and F335 increases [8]. In all of the LeuT 3D structures, however, approximately 20 Å of tightly packed helical regions effectively separate the substrate binding site from the cytoplasmic environment [4–8]. Thus, neither the crystal structures of LeuT nor the SERT homology models based on these structures reveal much information about how substrates are transported from the extracellular environment into the interiors of the cells. One possible way to gain more insight into the conformational mechanisms that take place in a transporter following the binding of either substrate or inhibitor may be by performing long molecular dynamics (MD) simulations.
To study ligand binding and SERT conformational changes upon ligand binding, the LeuT occluded structure (PDB id 2A65) [4] was used to generate a homology model of SERT, and 5-HT and ten other tryptamine derivatives, as well as the SSRI (S)-citalopram, were docked into the putative substrate binding pocket detected in the SERT model. Analysis of the docking results revealed two putative binding modes of the tryptamine derivatives and (S)-citalopram in SERT. Based on these docking results, one representative complex of SERT and 5-HT and (S)-citalopram in both binding modes was selected for MD simulations, in addition to the apo-SERT. The MD simulations were performed after embedding the SERT–ligand complexes in palmitoyloleoyl-phosphatidylcholine (POPC) lipid bilayers. The results from the MD simulations of the five SERT–(ligand)–POPC complexes showed that the putative substrate binding site had started to extend towards the intracellular parts of SERT during the MD simulation in one of the SERT–5-HT complexes (namely, the SERT–5-HTB complex). In the same complex, a vestibule extending from the cytoplasm towards the substrate binding site had started to form. Based on these results, we identified several amino acids that may play a role in the opening and closing of a vestibule reaching from the substrate binding site to the cytoplasm.
Methods
Homology modeling of SERT
The SERT (UniProtKB/Swiss-Prot accession number P31645 [14]) and the LeuT (PDB id 2A65) [4] amino acid sequences were aligned using ICM software (version 3.5) [15], and the alignment was adjusted to fit the published comprehensive alignment of NSS family members [3]. Based on this alignment, the homology model of SERT was constructed using the BuildModel macro of ICM [15]. The macro constructs the backbone of the target protein using the backbone conformation of the template in the aligned regions using core sections defined by the average Cα atom positions in these regions. The conformations of the side chains of amino acids that were identical for the template and the target structures were then transferred from the template to the target, whereas nonidentical side chains were assigned their most likely rotamer. For the loops with insertions or deletions between the template and target sequences, the macro performs a loop search of the PDB database, selecting loops with matching loop ends and a loop sequence that is as closed as possible. The loops are inserted into the model and the side chains are modified according to the model sequence and steric interactions with the surroundings of the model.
The SERT amino acids E78-T192 and W220-I608 were included in the homology model. These amino acids comprise the 12 putative transmembrane helices (TMs) and the intracellular and extracellular loops (ILs and ELs, respectively) connecting the transmembrane helices, except for parts of the large EL2 (amino acids 193–219). This loop segment was not included in the model as it is lacking in the LeuT template. Amino acids corresponding to the N-terminal (amino acids 1–77) and C-terminal (amino acids 609–630) regions of SERT were also not included in the model for the same reason.
The two sodium ion binding sites and one chloride binding site in the LeuT crystal structure [4] were copied to SERT after superimposing the LeuT crystal structure and the SERT model. A chloride ion was also added to the SERT homology model such that it occupied a position corresponding to the carboxylate carbon coordinates of LeuT glutamic acid at position 290 (corresponding to S372 in SERT), as suggested by Forrest [11] and Zomot [16].
Energy refinement of the SERT homology model was performed using the ICM RefineModel macro. This three-step macro performs (1) a side-chain conformational sampling using “Montecarlo fast” [17], (2) iterative annealing with tethers provided, and (3) a second side-chain sampling. The program module Montecarlo fast [17] samples the conformational space by performing iterations that consist of a random move followed by a local energy minimization. The complete energy is then calculated, and the iteration is accepted or rejected based on the energy and the temperature. In the annealing of the backbone (step 2), the tethers included are harmonic restraints that pull an atom in the model to a static point in space represented by a corresponding atom in the template.
The energy-refined SERT homology model was uploaded to the SAVES server for a structure quality check (http://nihserver.mbi.ucla.edu/Saves_3/). The Ramachandran plot provided by Procheck showed that the SERT homology model was a good-quality model; 96.6% of the non-glycine and non-proline amino acids were in the favored regions, whereas 3.4% (12 amino acids) were in additional allowed regions. Of these 12 amino acids, one amino acid, D98, was located in the putative substrate binding area. This amino acid is important for substrate and inhibitor binding to SERT [10, 12, 18–20], and was located in an unwound region of TM1. However, this location gives D98 more freedom to rotate, and hence explains its location in additionally allowed regions of the Ramachandran plot.
Ligand docking
To detect possible binding pockets in the SERT structure, the ICM PocketFinder macro was used (default tolerance level of 4.6). The algorithm uses a transformation of the Lennard–Jones potential calculated from a three-dimensional protein structure and does not require any knowledge about a potential ligand molecule; i.e., it is based solely on protein structure [21].
5-HT and ten other tryptamine derivatives (tryptamine, 4-hydroxytryptamine (4-HT), 7-methyltryptamine (7-MT), 2-methylserotonin (2-MT), 5-methoxy-3-(1,2,5,6-tetrahydro-4-pyridinyl)-1 H-indole (RU24969), N-isopropyltryptamine (NIT), 5-methoxy-N-isopropyltryptamine (5MNIT), 7-benzyloxytryptamine (7-BT), 5,6,7-trihydroxytryptamine and serotonin o-sulfate (Table 1) were constructed using the ChemDraw option of ICM. Default ECEPP/3 partial charges were assigned to the protonated forms of the ligands [22], and the compounds were docked using the batch docking method of ICM. RU24969 was also docked in its unprotonated state. The SSRI [(S)-1-[3-(dimethylamino)propyl]-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5 carbonitrile; (S)-citalopram] (Table 1) was constructed using ChemDraw and docked into the same binding site as the tryptamine derivatives, as experimental studies indicate that (S)-citalopram is a competitive 5-HT inhibitor [18]. The ligands were docked using a semi-flexible docking protocol where SERT was kept rigid but the ligands flexible.
The poses of each ligand were clustered and compared with the clusters of the other ligands. This analysis led to the identification of two putative ligand positions for both the tryptamine derivatives and (S)-citalopram. One representative from each of the two clusters of 5-HT (representing the tryptamine derivatives) and (S)-citalopram were selected for MD simulations.
Molecular dynamics simulation
The automated CHARMM-GUI membrane builder tool [23] was used for the generation of a palmitoyloleoylphosphatidylcholine (POPC) lipid bilayer around the five SERT–(ligand) complexes selected after docking. The pre-orientated LeuT structure [4] from the Orientations of Proteins in Membranes (OPM) database [24] was used to orient the SERT model in the membrane by superimposing the LeuT and SERT. An unequilibrated lipid bilayer was generated using the replacement method, in which SERT was packed with lipid-like spheres whose positions then were used to place randomly chosen POPC lipid molecules from a lipid library composed of 2000 different conformations of lipids generated by MD simulations of pure lipid bilayers. The dimensions of the entire SERT–(ligand)–POPC molecular system was approximately 100×100×100 Å, including 1 Å extra added in each direction in order to introduce space between the boundary of the system and the boundary atoms of the simulation cell. One hundred fifteen lipids were included in the outer bilayer and 121 in the inner bilayer. Water molecules (TIP3) and K+ and Cl− ions were then added by the membrane builder tool to fully solvate the system. In total, each of the five complexes consisted of approximately 98,000 atoms.
The NAMD scalable MD simulator (versions 2.6 and 2.7b1) [25] was used to equilibrate the systems and perform the production runs. The MD simulations were run using 64 processors on the Stallo supercomputer at the University of Tromsø, Norway, using Chemistry at HARvard Molecular Mechanics (CHARMM) force fields. The CHARMM par_all27_prot_lipidNBFIX parameter file, which includes the CHARMM22/CMAP force field [26, 27] for the protein and the CHARMM27 force field [28, 29] for lipids, was used. For the complexes containing 5-HT or (S)-citalopram, the CHARMM36 general force field for small molecule drug design (CGenFF v. 2a3 [30]) was included, manually adding force field angle and dihedral parameters that are not included in CGenFF v. 2a3 [30]. To allow the large volume fluctuations that are typical of the initial dynamics of a new system in an NPT ensemble, a margin of 5 was used during the equilibration steps, which was reduced to 2 during the production runs [25]. During the simulations, Nosé–Hoover–Langevin dynamics were used to simulate the NPT ensemble. This method combines the Nosé–Hoover constant pressure method with piston fluctuation control implemented using Langevin dynamics by coupling the piston to a heat bath. A damping constant of 10/langevinPistonDecay was used during the equilibration steps, which was reduced to 1/langevinPistonDecay during the production runs. The langevinPistonDecay (50 fs) was set to be smaller than langevinPistonPeriod (200 fs) to ensure that harmonic oscillations in the periodic cell were overdamped. The target pressure was set at 1.01325 bar (atmospheric pressure at sea level), and group-based pressure (useGroupPressure) was used to control the periodic cell fluctuations, as the atom-based pressure has more high-frequency noise. In addition, a flexible cell (useFlexibleCell) was used, allowing the height, length, and width of the cell to fluctuate independently during the simulation, which is very useful for anisotropic systems such as membranes.
The equilibration of the five SERT–(ligand)–POPC complexes consisted of three steps during which the system was gradually released. During steps (1) and (2), harmonic constraints of 1 kcal mol−1 Å−2 were specified in the PDB beta field of each atom to be constrained. In order to induce the appropriate order of the fluid-like bilayer, all atoms except the lipid tail atoms were constrained during step (1), and lipids, water and ions were permitted to adapt to the structure of the protein. During step (2), only protein atoms were constrained, whereas the whole system was released during step (3). During step (1), 10,000 steps of conjugate gradient energy minimization were performed, followed by 10,000 steps (10 ps) of system heating to 300 K under constant temperature control and 500,000 steps (0.5 ns) of MD. During steps (2) and (3), only 10,000 steps of conjugate gradient minimization followed by 500,000 steps (0.5 ns) of MD were performed. In total, 30,000 steps of conjugate gradient minimization, 10 ps of heating and 1.5 ns of MD simulations were run to equilibrate the system. To confirm that the systems stabilized during equilibration, the RMSD from the starting structure was monitored during each simulation using the molecular dynamics (VMD) viewer version 1.8.6 [31]. Finally, the equilibration phases of the SERT–5-HT binding modes A and B and the (S)-citalopram binding modes A and B, as well as SERT alone, were followed by 22, 21, 32, 23 and 25 ns MD simulations, respectively. The production simulations were performed at 300 K. Following the production runs, VMD [31] was used to generate average structures of each complex based on the last 10 ns of each simulation, and ICM PocketFinder [21] was used to detect possible pockets in the average structures. Based on the these analyses, the SERT–5-HTB complex MD simulation was prolonged to 49 ns.
Results
Homology modeling
The constructed homology model consisted of 12 TMs, among which TMs 1–5 and 6–10 were arranged with a pseudo-twofold axis in the membrane plane, as for LeuT [4]. Three possible binding pockets were identified by ICM PocketFinder in the SERT homology model: one in the region corresponding to the LeuT substrate binding site, and two extracellular pockets which were separated from the putative substrate binding pocket by the side chains of Y176 and F335, the aromatic amino acids of the extracellular gate. In LeuT [4], only one pocket was detected in this extracellular region, as EL4 in LeuT is missing three amino acids at the tip of EL4 as compared to SERT [3] (results not shown).
ICM PocketFinder [21] identified a binding pocket that corresponded to the substrate binding site of LeuT [4]. Experimental data on SERT and the X-ray structure of LeuT also suggest that the substrate binding site of SERT and LeuT are in the same region [10, 12, 20, 32–34], halfway across the membrane bilayer within the TMs. This location is also consisted with the alternate access theory [13]. Amino acids from four TMs contribute to the binding pocket detected by ICM PocketFinder, namely from TM1 (Y95, D98, G100), TM3 (I172, A173, Y176), TM6 (F335, S336, G338, F341, V343) and TM8 (S438, T439, G442). An important feature of the detected binding pocket is the deviation from regular helical structure in the unwound regions of TM1 (A96–D98) and TM6 (G338–G342). A similar deviation is observed in corresponding regions of the X-ray structure of LeuT. In the unwound regions, the main-chain carbonyl oxygen and amide nitrogen atoms are exposed such that they can easily take part in direct hydrogen-bonding interactions with ligands and coordinate ions.
The substrate binding pocket detected by ICM PocketFinder could be divided into three subpockets based on the main properties of amino acids involved. The first subpocket, the hydrophobic subpocket, was located towards the intracellular end of the binding site and was surrounded by the side chains of A169 (TM3), A173 (TM3), V343 (TM6), and G442 (TM8). The side chain of I172 (TM3) was positioned such that it could participate in forming the hydrophobic subpocket but also separate the hydrophobic subpocket from an aromatic. The aromatic subpocket consisted of the side chains of the two aromatic amino acids of the extracellular gate, Y176 (TM3) and F335, and F341 located in the unwound region of TM6. The third subpocket, the ionic subpocket, was located in the vicinity of D98 (TM1).
Analysis of the docking results
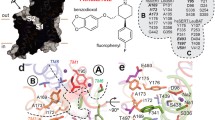
The docking of 5-HT and ten other tryptamine derivatives and (S)-citalopram indicated two possible binding modes of the compounds, designated SERT–5-HTA, SERT–5-HTB, SERT–(S)-citalopramA and SERT–(S)-citalopramB, respectively (Fig. 1). The SERT–5-HT binding modes represent the binding poses of all tryptamine derivatives. In both the SERT–5-HTA and SERT–5-HTB binding modes, 5-HT occupied the ionic and hydrophobic—but not the aromatic—subpockets of the binding site. The protonated amine of 5-HT was located near the D98 carboxyl side chain in both modes, which is in accordance with experimental data [10, 12, 19, 20]. The two binding modes of 5-HT differ in the orientation of the indole ring nitrogen and the orientation of the 5 position (Fig. 1). In the SERT–5-HTA binding mode, the indole ring nitrogen was found between Y95 and F341, whereas the 5 position was pointing towards Y176, S438 and T439. In the SERT–5-HTB binding mode, however, the indole ring was flipped 180° compared to binding mode A, and the indole nitrogen group was pointing towards the aromatic side chains of Y176 and S438, and the 5 position towards A169 and F341 (Fig. 1). Interestingly, similar binding modes of 5-HT to the SERT–5-HTA and SERT–5-HTB binding modes have also been described by other groups [10, 12, 35]. Predictions of the 5-HT–SERT binding energies for the two binding modes using the calcBindingEnergy macro of ICM [36] showed that the poses represented by the SERT–5-HTA complex had binding energies in the range −5.7 to −13.8 kcal mol−1 (average −10.0 kcal mol−1), while poses represented by the SERT–5-HTB complex had binding energies in the range −4.8 to −10.7 kcal mol−1 (average −8.1 kcal mol−1).

Ligand binding modes detected through docking. a SERT–5-HTA binding mode, b SERT–5-HTB binding mode, c SERT–(S)-citalopramA binding mode, and d SERT–(S)-citalopramB binding mode. The side chains of amino acids Y95, D98 and I172 and the binding pocket detected by ICM PocketFinder (red wire representation) are shown. Color coding of atoms in amino acids: red oxygen, blue nitrogen, gray carbon and hydrogen. Color coding of ligands: red oxygen, blue nitrogen, yellow carbon, gray hydrogen
In the SERT–(S)-citalopramA binding mode (Fig. 1), (S)-citalopram occupied all three subpockets of the putative substrate binding site. The amine moiety of (S)-citalopram was located in the ionic subpocket close to D98, whereas the cyanophthalane and fluorophenyl moieties were located in the hydrophobic (in close proximity to A169, A173, V343 and G442) and aromatic subpockets (pointing towards F335), respectively. The oxygen moiety of (S)-citalopram was pointing in the direction of Y95 (Fig. 1). In comparison, the cyanophthalane and amine moieties of (S)-citalopram in the SERT–(S)-citalopramB binding mode were also found in the hydrophobic and ionic subpockets, respectively, in a very similar location to that in the SERT–(S)-citalopramA binding mode. However, the fluorophenyl moiety of (S)-citalopram in this binding mode was found to be juxtaposed in-between the side chains of Y95 and S438, and the oxygen moiety was pointing in the direction of Y176 (Fig. 1). The prediction of binding energies using the calcBindingEnergy macro of ICM [36] showed that poses represented by the SERT–(S)-citalopramA complex had binding energies in the range −7.4 to −19.1 kcal mol−1 (average −14.7 kcal mol−1), while those represented by the SERT–(S)-citalopramB complex had binding energies in the range −12.7 to −19.7 kcal mol−1 (average −16.4 kcal mol−1).
Molecular dynamics simulations
In order to study possible conformational changes of SERT upon the binding of 5-HT (substrate) and (S)-citalopram (inhibitor), more than 20 ns of MD simulations were performed for each system: one representative SERT–ligand complex from each of the binding modes detected as well as apo-SERT were embedded in POPC lipid bilayers, followed by system equilibration and longer MD simulations. The average structures of each of the five complexes were then generated based on the last 10 ns of the production runs, and ICM PocketFinder was used to detect possible pockets that had formed in SERT during the production runs.
Interestingly, in the average structure of the SERT–5-HTB binding mode, the substrate binding pocket began to elongate towards the cytoplasm, and another pocket started to form that extended from the cytoplasm up towards the elongated substrate binding pocket during the MD simulation (Fig. 2). Our results showed that in the average structure of SERT–5-HTB, only a narrow stretch of TMs 6 and 8, in addition to intracellular loop 1 (IL1), separated the two pockets and prevented access from the substrate binding site to cytoplasm (Fig. 3). The other simulations also changed the size of the substrate binding site and induced other pockets to form; however, intracellular vestibules similar to that generated in the SERT–5-HTB complex were not observed in any of the other average structures (results not shown). Based on these observations, the simulation of the SERT–5-HTB complex was prolonged to 49 ns. The prolongation indicated that the pocket extending from the cytoplasm up towards the elongated substrate binding pocket was also maintained during 21 to 49 ns of the MD simulation.

SERT structures. a Initial SERT structure and b the average SERT–5-HTB structure generated based on the last 10 ns of the MD simulation. “Intra-structural” pockets detected by ICM PocketFinder are shown. The putative substrate binding pocket is represented as red wire

a Intracellular view of the average SERT–5-HTB structure. SERT Cα carbon atoms are shown in gray cylindrical representation. For clarity, amino acids 148–160, 338–350 and 444–453 are shown in blue. The putative substrate binding site is displayed as red wire. Amino acids that are proposed to play a role in the opening of a vestibule extending from the putative substrate binding site (red wire representation) to the cytoplasm are shown as xstick. b Close-up of a with residues in xstick. Green lines show interactions formed during the simulation; red line shows an interaction broken during simulation
The 5-HT in the average SERT–5-HTB structure (12–21 ns) was slightly shifted compared with the initial structure (Fig. 4). Superimposition of the structure of SERT prior to MD and the average structure of the SERT–5-HTB complex showed that the hydroxyl oxygen atom of 5-HT was located closer to the Y95 (TM1) hydroxyl group. The distance before MD was 4.1 Å, while the distance in the average structure was 3.4 Å (range 1.9–5.5 Å). 5-HT was also located 1.7 Å closer to the cytoplasmic side than before MD. The distance between the G338 (TM6) backbone oxygen and the Y95 (TM1) hydroxyl group also increased slightly, from 1.8 Å to 2.1 Å in the average structure (range 2.0–3.0 Å), indicating that TMs 1 and 6 had begun to move further apart as well (Fig. 4). Prolongation of the MD indicated that these distances did not change much during 21–49 ns of MD. The distance between the 5-HT hydroxyl group and the hydroxyl group of Y95 varied between 2.3 and 5.3 Å, while the distance between the G388 backbone oxygen and the Y95 hydroxyl group varied between 1.8 and 2.7 Å.

Comparison of the 5-HT binding mode in the initial SERT–5-HTB complex (gray) and that in the average SERT–5-HTB structure generated based on the last 10 ns of MD (orange). Atomic distances (Å) are shown as dotted lines. For clarity, selected hydroxyl oxygen atoms on 5-HT, Y95 and G338 are colored red
The observation that only some residues block the access from the putative substrate binding site to the cytoplasm prompted us to look for amino acids in the unwound region of TM6, in TM8, and in IL1 of SERT that may have interacted with amino acids in other regions of SERT and contributed to the formation of the emerging vestibule. We found G340 in TM6 and E444, D452 and E453 in TM8, as well as R152 and K153 in IL1 very interesting in this respect. The distances between these residues and their interaction partners in the structure of SERT before the MD simulations and in the average structures generated following the MD simulations were thus measured and compared (Table 2).
We also noted that the cytoplasmic part of TM3 (K159–I168) had unwound during the MD simulation and had thus become more flexible. The unwinding may have played a role in the opening of the vestibule; however, this unwinding was seen in all average structures and may be an artifact of poor force field representation of protein–protein, protein–solvent and solvent–lipid interactions. Using CHARMM force fields and NPT for simulations in a tensionless ensemble may lead to the condensation of the bilayer to a near gel-like state, which may influence the protein structure and result in incorrect predictions if the lateral density of lipids increases beyond a liquid crystalline state [37]. The unwinding may also be a result of structural differences between SERT and LeuT in this region [3].
Structural differences between SERT and LeuT in IL1 may explain the unwinding of the α-helical structure in IL1 that was present in the initial structure of SERT, just as in LeuT [4], but not in any of the average structures generated following the MD simulations. The homology between SERT and LeuT in this region is very low, with only one identical amino acid (I154, SERT numbering) [3], and the presence of an α-helical structure in IL1 of SERT is thus questionable.
Discussion
Homology modeling and docking
The homology modeling approach is a valuable tool for investigating protein structures when experimental structures are lacking. Homology models are useful for predicting ligand potency and specificity through the use of different docking approaches, and high-quality homology models have also been used in the study of conformational changes using MD simulations [38]. In the present study, 5-HT and ten other tryptamine derivatives (SERT substrates) and the SSRI (S)-citalopram were docked into the putative substrate binding site of a SERT homology model, and possible conformational changes of SERT upon ligand binding were studied by MD simulations.
The accuracy of homology models depends on three factors: the sequence identity and functional similarity between the template and target proteins; the amino acid sequence alignments between the template and the targets; and the resolution at which the crystal structure of the template protein was resolved. For membrane proteins in general, sequence identities between template and target proteins of 50% have been found to yield membrane homology models with a Cα-RMSD of approximately 1 Å from the template structure in the transmembrane regions, assuming that the template structure has been solved at a resolution of 3.5 Å or better [39]. Sequence identities of 30% or more are, for most membrane proteins, predicted to yield acceptable homology models with a Cα-RMSD of approximately 2 Å in the TM regions [39].
The sequence identity between LeuT and SERT is approximately 50% in the putative substrate binding site detected by the ICM PocketFinder. In contrast, the overall sequence identity between the transporters is less than 20%, but it rises to approximately 35% in TMs that are predicted to be directly involved in substrate binding (i.e., TMs 1, 3, 6 and 8). LeuT is considered a good template for generating homology models of SERT that can be used for ligand docking and molecular dynamics. Actually, due to the topological restrictions provided by the hydrophobic membrane environment surroundings, membrane proteins such as SERT actually have more limited ways of folding than water-soluble proteins, which may suggest that membrane protein homology models are more accurate than homology models of water-soluble proteins at the same level of sequence identity [39]. This also thus supports the generation of acceptable homology models of not only the SERT substrate binding site but the whole structure using LeuT as a template.
Our docking results suggest two different ways 5-HT and the other tryptamine derivatives may bind in SERT: the SERT–5-HTA and SERT–5-HTB binding modes (Fig. 1). In both of these binding modes, the positively charged amine moiety of 5-HT was in the vicinity of the negatively charged D98 side chain, and the C6 position of the indole ring was located close to A173 at the other end of the molecule; however, the indole nitrogen moiety pointed in different directions in the two binding modes. Interestingly, similar binding modes of 5-HT to the SERT–5-HTA and SERT–5-HTB binding modes have also been obtained through docking and experimental studies by other groups [10, 12, 35]. Celik et al. [10] found that the C5 and C7 positions of 5-HT should be located in hydrophilic and hydrophobic pockets of SERT, and that the 5 hydroxyl moiety of 5-HT was in the vicinity of T439 (TM8) [10]. Though the C5 and C7 moieties of 5-HT in both the SERT–5-HTA and SERT–5-HTB binding modes described here are located in such regions, only the localization of C5 of 5-HT in the SERT–5-HTA binding mode was found in the vicinity of T439. In another study, however, 5-HT in a similar binding mode to the SERT–5-HTB binding mode showed good correlation with experimental data and was also found to best describe the cross-species sensitivities reported in support vector machine (SVM) sensitivity maps generated for the human and Drosophila melanogaster serotonin transporters [12]. This binding mode was also suggested by Jørgensen et al. [35].
Our results show that the size of the putative substrate binding site detected in this structure of SERT was relatively small and not optimal for the docking of larger compounds such as (S)-citalopram. Nonetheless, the binding mode of (S)-citalopram has recently been studied by docking into occluded SERT homology models and by experimental site-directed mutagenesis [18]. Andersen et al. [18] found that the fluorophenyl moiety of (S)-citalopram was located near I172, A173 and N177, whereas the cyanophthalane moiety was in proximity to V343. Though the cyanophthalane moiety of (S)-citalopram in both binding modes in the present study was in the vicinity of V343, only the fluorophenyl of (S)-citalopram in the SERT–(S)-citalopramA binding mode was in the vicinity of I172 (Fig. 1). A similar (S)-citalopram binding mode to the SERT–(S)-citalopramA binding mode has also been used as initial binding mode in another MD study in SERT [35].
Our docking indicated that the tryptamine derivatives do not interact with SERT in the aromatic subpocket of the binding pocket, whereas (S)-citalopram does. A possible mechanism of action of inhibition by (S)-citalopram may therefore be that (S)-citalopram interferes with the closure of the extracellular gating residues Y176 and F335, stabilizing SERT in an outward-facing conformation, thereby hindering conformational changes needed for transport to occur. A similar mechanism of inhibition has recently been suggested for TCAs [40].
Molecular dynamics simulations
In order to gain insights into SERT conformational changes that may take place upon ligand binding, one representative ligand orientation from each of the two possible binding modes of 5-HT (representing the tryptamine derivatives) and (S)-citalopram, as well as the apo-SERT structure, were selected for MD simulations in POPC lipid bilayers. The simulations were run for 22 ns (SERT–5-HTA), 49 ns (SERT–5-HTB), 32 ns (SERT–(S)-citalopramA), 23 ns (SERT–(S)-citalopramB) and 25 ns (apo-SERT), and average structures of each of the five MD simulations were generated and used to analyze the results. Average structures may represent unphysical states of SERT that may not exist. However, the present average structures were based on the last 10 ns of the MD simulation, where energetically favorable and structural stable SERT–(ligand)–POPC complexes were obtained. The average structures used were thus considered to be representative of the most densely populated conformations during this period of the simulation.
The substrate 5-HT is expected to cause a different conformational change of SERT than inhibitors such as (S)-citalopram, as the former compound is transported whereas the latter inhibits transport. In order to visualize such conformational changes, the ICM PocketFinder was used to detect pockets in the five average structures. In the average structure from SERT-5HTB binding mode simulation, the pockets detected showed that a vestibule had started to emerge that extended from the putative substrate binding site towards the cytoplasm (Fig. 2). The results suggested that the continued rearrangement of the unwound regions of TM6, TM8 and IL1 relative to one another may open a pathway from the substrate binding site to the cytoplasm (Fig. 3). A similar vestibule was not observed in any of the other simulations (results not shown).
A pocket extending from the cytoplasm up towards the substrate binding pocket was formed during the MD simulation of the SERT–5-HTB complex. A corresponding pocket was not formed during MD of the SERT–5-HTA complex. Based on these observations, we also examined whether the position of 5-HT changed during the simulation of the SERT–5-HTB complex. By superimposing the initial structure of SERT on the average SERT–5-HTB structure (12–21 ns), we found that the 5-HT hydroxyl group was located closer to the Y95 (TM1) hydroxyl group at the cytoplasmic end of the binding pocket in the average SERT–5-HTB structure. In addition, the atomic distance between Y95 (TM1) and G338 (TM6) was slightly increased (Fig. 4). Prolonging the MD simulation up to 49 ns showed that these distances were maintained between 21 and 49 ns of MD simulation, and additional changes in SERT structure or in 5-HT position were not seen.
The hydroxyl group of Y95 (TM1) and the backbone oxygen atom of G338 in the unwound region of TM6 were within hydrogen-bonding distance in the initial structure of SERT, and this interaction might play a role in keeping the translocation pathway closed. Our results thus suggest that one of the first steps in 5-HT translocation is the formation of a hydrogen bond between the 5-OH of 5-HT and Y95 (TM1), which may sever the hydrogen bond between Y95 (TM1) and G338 (TM6). In another study, the mutation of G338 to cysteine (G338C) was shown to stabilize SERT in an outward-facing conformation [33]. The transport activity of the G338C mutant was less than 5% of the wild-type transport activity; however, transport could partially be restored by simultaneously mutating Y95 to phenylalanine (Y95F), which indicates that Y95 (TM1) and G338 (TM6) cannot be hydrogen bonded for 5-HT transport to occur [33].
The amino acids in TM6 that separated the putative substrate binding site from the cytoplasmic vestibule were located in the unwound region of TM6, which in the initial SERT structure consisted of G338, P339, G340, F341 and G342, but in the average SERT–5-HTB structure also contained two more amino acids, S336 and L337. The unwinding of the latter amino acids is in agreement with a study suggesting that amino acids 334–337 in SERT are in an unwound region based on aqueous accessibility data [33]. This region contains several glycine residues [3] and is thus expected to be very flexible: one study shows that even the conservative mutations of G338 and G342 to alanine (G338A and G342A, respectively) cause reductions in 5-HT transport of approximately 28% and 10%, respectively, as compared to the wild type [33].
The transmembrane helix closest to TM6 in the model was TM2. Thus, an interaction between the unwound region of TM6 and amino acids in TM2 might contribute to opening up the binding site towards the intracellular region by pulling the flexible unwound part of TM6 towards TM2. We observed that a hydrogen bond was present between the backbone of G340 (unwound region of TM6) and the side chain of E136 (TM2), as in LeuT [4]. Our results show that the distance between the backbone nitrogen of G340 and the E136 side chain did not change significantly during the MD simulation of the SERT–5-HTB complex (Table 2); however, superimposing the average structure on the initial SERT structure showed that the G340 backbone nitrogen atom and the E136 carboxyl carbon atoms shifted 2.5 Å during the simulation (results not shown). Hence, though the distance between G340 and E136 remains constant during the MD simulation, the unwound TM6 region and TM2 had moved 2.5 Å in the same direction, away from the putative substrate binding site. An ionic interaction between another TM2 amino acid, R144, and E78 in the N-terminus also formed, and may have contributed to the joint movement of TMs 2 and 6. E136 (TM2) is conserved among the Na+-dependent NSS transporters [3], and has been shown to be very important for transport in SERT: a conservative mutation of this glutamic acid to aspartic acid (E136D) causes a reduction in SERT transport, and mutations to alanine or glutamine (E136A, E136Q) inhibit transport [41]. The atomic distance between R144 (TM2) and E78 (N-terminus) decreased from 7 Å in the initial structure of SERT to 1.9 Å in the average structure of SERT–5-HTB (Table 2).
In TM8, three amino acids were found to be particularly interesting with respect to opening an intracellular vestibule from the putative substrate binding site to the cytoplasm: namely E444, D452 and E453. E444 (TM8) was located in close proximity to the substrate binding site, and during all MD simulations an ionic interaction between E444 (TM8) and R462 (TM9) was formed (Table 2). D452 and E453 were located at the cytoplasmic end of the TM8. During the MD simulation of the SERT–5-HTB complex, we observed that the distance between E453 (TM8) and R152 (IL1) increased whereas the distance between D452 (TM8) and K153 (IL1) decreased, thus changing the conformation of this long loop. The importance of R152 for transport is in agreement with a recent study in mouse SERT showing that the G39/K152 phenotype has reduced transport in comparison with the wild type (E39/R152 phenotype) [42].
Very interestingly, we observed that during the MD simulation of SERT–5-HTB, an interaction between R152 (IL1) and E508 (TM10) developed. In the initial structure of SERT, the atomic distance between these residues was >17 Å, while the distance decreased to only 2.7 Å in the average SERT–5-HTB structure (Table 2). Furthermore, this interaction was not formed in any of the other MD simulations (Table 2). E508 is one of a few amino acids in TM10 that are fully conserved between SERT and LeuT [3]. Interestingly, E508 (TM10) was also located in the region of E136 (TM2) in SERT, and it is suggested that this amino acid interacts with G340 in the unwound region of TM6 (see above); it is also known to be important for transport in SERT [41].
Summary
Our MD simulations indicate that the SERT–5-HTB binding mode and not the SERT–5-HTA binding mode induces conformational changes in SERT that may be associated with substrate translocation. The simulations suggest that substrate translocation may involve forming and breaking ionic interactions between TM6, TM8 and IL1 and their interaction partners. Although our observations are in agreement with experimental studies, the suggested mechanism is hypothetical, as it is based solely upon theoretical calculations using a homology-based model.
The simulations may indicate that the formation of a hydrogen bond between Y95 in TM1 and 5-HT causes a hydrogen bond between Y95 and G338 in TM6 to be broken, enabling the unwound region of TM6 to move away from the substrate binding site and transport to begin. The formation of an ionic interaction between R144 (TM2) and E78 (N-terminus) and the interaction between G340 (unwound region of TM6) and E136 (TM2) then cause TM6 to move away from the putative substrate binding site. The movements of E136 (TM2) also affect the nearby amino acid E508 (TM10), causing it to interact with R152 in IL1, thus changing the conformation of this loop. Simultaneously, an ionic interaction between E444 (TM8) and R462 (TM9), located close to the putative substrate binding site, is formed. The interaction between E453 in the cytoplasmic part of TM8 and R596 in TM12 may also contribute to relocating TM8 away from the vestibule. The formation of an ionic interaction between E78 in the N-terminus and R144 in TM2, and the subsequent movement of TM2, may also weaken the interaction between the N-terminus and TM8, as illustrated by the increase in the R79–D452 distance (Table 2).
References
Saier MH Jr (2000) A functional-phylogenetic classification system for transmembrane solute transporters. Microbiol Mol Biol Rev 64:354–411
Chen NH, Reith ME, Quick MW (2004) Synaptic uptake and beyond: the sodium- and chloride-dependent neurotransmitter transporter family SLC6. Pflugers Arch 447:519–531
Beuming T, Shi L, Javitch JA, Weinstein H (2006) A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol Pharmacol 70:1630–1642
Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E (2005) Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 437:215–223
Singh SK, Yamashita A, Gouaux E (2007) Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature 448:952–956
Zhou Z, Zhen J, Karpowich NK, Goetz RM, Law CJ, Reith ME, Wang DN (2007) LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science 317:1390–1393
Zhou Z, Zhen J, Karpowich NK, Law CJ, Reith ME, Wang DN (2009) Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nat Struct Mol Biol 16:652–657
Singh SK, Piscitelli CL, Yamashita A, Gouaux E (2008) A competitive inhibitor traps LeuT in an open-to-out conformation. Science 322:1655–1661
Chothia C, Lesk AM (1986) The relation between the divergence of sequence and structure in proteins. EMBO J 5:823–826
Celik L, Sinning S, Severinsen K, Hansen CG, Moller MS, Bols M, Wiborg O, Schiott B (2008) Binding of serotonin to the human serotonin transporter. Molecular modeling and experimental validation. J Am Chem Soc 130:3853–3865
Forrest LR, Tavoulari S, Zhang YW, Rudnick G, Honig B (2007) Identification of a chloride ion binding site in Na+/Cl−-dependent transporters. Proc Natl Acad Sci USA 104:12761–12766
Kaufmann KW, Dawson ES, Henry LK, Field JR, Blakely RD, Meiler J (2009) Structural determinants of species-selective substrate recognition in human and Drosophila serotonin transporters revealed through computational docking studies. Proteins 74:630–642
Jardetzky O (1966) Simple allosteric model for membrane pumps. Nature 211:969–970
Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, Yeh LS (2004) UniProt: the Universal Protein knowledgebase. Nucleic Acids Res 32:D115–119
Abagyan RT, Kuznetsov D (1994) ICM—a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J Comput Chem 15:488–506
Zomot E, Bendahan A, Quick M, Zhao Y, Javitch JA, Kanner BI (2007) Mechanism of chloride interaction with neurotransmitter: sodium symporters. Nature 449:726–730
Abagyan R, Totrov M (1994) Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol 235:983–1002
Andersen J, Olsen L, Hansen KB, Taboureau O, Jorgensen FS, Jorgensen AM, Bang-Andersen B, Egebjerg J, Stromgaard K, Kristensen AS (2010) Mutational mapping and modeling of the binding site for (S)-citalopram in the human serotonin transporter. J Biol Chem 285:2051–2063
Andersen J, Taboureau O, Hansen KB, Olsen L, Egebjerg J, Stromgaard K, Kristensen AS (2009) Location of the antidepressant binding site in the serotonin transporter: importance of Ser-438 in recognition of citalopram and tricyclic antidepressants. J Biol Chem 284:10276–10284
Barker EL, Moore KR, Rakhshan F, Blakely RD (1999) Transmembrane domain I contributes to the permeation pathway for serotonin and ions in the serotonin transporter. J Neurosci 19:4705–4717
An J, Totrov M, Abagyan R (2005) Pocketome via comprehensive identification and classification of ligand binding envelopes. Mol Cell Proteomics 4:752–761
Nemethy G, Gibson KD, Palmer KA, Yoon CN, Paterlini G, Zagari A, Rumsey S, Scheraga HA (1992) Energy parameters in polypeptides. 10. Improved geometrical parameters and nonbonded interactions for use in the Ecepp/3 algorithm, with application to proline-containing peptides. J Phys Chem 96:6472–6484
Jo S, Kim T, Im W (2007) Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE 2:e880
Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI (2006) OPM: orientations of proteins in membranes database. Bioinformatics 22:623–625
Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802
Mackerell AD Jr, Bashford D, Bellot M, Dunbrack RL Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Leu FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reither WE III, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102:3586–3616
Mackerell AD Jr, Feig M, Brooks CL III (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem 25:1400–1415
Feller SE, MacKerell AD (2000) An improved empirical potential energy function for molecular simulations of phospholipids. J Phys Chem B 104:7510–7515
Feller SE, Gawrisch K, MacKerell AD Jr (2002) Polyunsaturated fatty acids in lipid bilayers: intrinsic and environmental contributions to their unique physical properties. J Am Chem Soc 124:318–326
Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD Jr (2009) CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31:671–690
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14(33–38):27–38
Adkins EM, Barker EL, Blakely RD (2001) Interactions of tryptamine derivatives with serotonin transporter species variants implicate transmembrane domain I in substrate recognition. Mol Pharmacol 59:514–523
Field JR, Henry LK, Blakely RD (2010) Transmembrane domain 6 of the human serotonin transporter contributes to an aqueously accessible binding pocket for serotonin and the psychostimulant 3,4-methylene dioxymethamphetamine. J Biol Chem 285:11270–11280
Walline CC, Nichols DE, Carroll FI, Barker EL (2008) Comparative molecular field analysis using selectivity fields reveals residues in the third transmembrane helix of the serotonin transporter associated with substrate and antagonist recognition. J Pharmacol Exp Ther 325:791–800
Jorgensen AM, Tagmose L, Jorgensen AM, Bogeso KP, Peters GH (2007) Molecular dynamics simulations of Na+/Cl−-dependent neurotransmitter transporters in a membrane-aqueous system. Chem Med Chem 2:827–840
Schapira M, Totrov M, Abagyan R (1999) Prediction of the binding energy for small molecules, peptides and proteins. J Mol Recognit 12:177–190
Benz RW, Castro-Roman F, Tobias DJ, White SH (2005) Experimental validation of molecular dynamics simulations of lipid bilayers: a new approach. Biophys J 88:805–817
Vashisth H, Abrams CF (2010) All-atom structural models for complexes of insulin-like growth factors IGF1 and IGF2 with their cognate receptor. J Mol Biol 400:645–658
Forrest LR, Tang CL, Honig B (2006) On the accuracy of homology modeling and sequence alignment methods applied to membrane proteins. Biophys J 91:508–517
Sinning S, Musgaard M, Jensen M, Severinsen K, Celik L, Koldso H, Meyer T, Bols M, Jensen HH, Schiott B, Wiborg O (2009) Binding and orientation of tricyclic antidepressants within the central substrate site of the human serotonin transporter. J Biol Chem 285:8363–8374
Korkhov VM, Holy M, Freissmuth M, Sitte HH (2006) The conserved glutamate (Glu136) in transmembrane domain 2 of the serotonin transporter is required for the conformational switch in the transport cycle. J Biol Chem 281:13439–13448
Carneiro AM, Airey DC, Thompson B, Zhu CB, Lu L, Chesler EJ, Erikson KM, Blakely RD (2009) Functional coding variation in recombinant inbred mouse lines reveals multiple serotonin transporter-associated phenotypes. Proc Natl Acad Sci USA 106:2047–2052
Acknowledgments
This work was supported by a grant from the Nevronor program of the Research Council of Norway (project 176956), by the Polish–Norwegian Research Fund (grant PNRF-103-A1-1/07), and by CPU hours from NOTUR (Norwegian Metacenter for Computational Science). NAMD was developed by the Theoretical and Computational Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana-Champaign.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Mari Gabrielsen is a fellow of the Ph.D. school in Molecular and Structural Biology (MSB) at the University of Tromsø, Norway.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Gabrielsen, M., Ravna, A.W., Kristiansen, K. et al. Substrate binding and translocation of the serotonin transporter studied by docking and molecular dynamics simulations. J Mol Model 18, 1073–1085 (2012). https://doi.org/10.1007/s00894-011-1133-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-011-1133-1