
Abstract
The harmonic and anharmonic frequencies of fundamental vibrations in formaldehyde and water were successfully estimated using the B3LYP Kohn-Sham limit. The results obtained with polarization- and correlation-consistent basis sets were fitted with a two-parameter formula. Anharmonic corrections were obtained by a second order perturbation treatment (PT2). We compared the performance of the PT2 scheme on the two title molecules using SCF, MP2 and DFT (BLYP, B3LYP, PBE and B3PW91 functionals) methods combined with polarization consistent pc-n (n = 0, 1, 2, 3, 4) basis sets, Dunning’s basis sets (aug)-cc-pVXZ where X = D, T, Q, 5, 6 and Pople’s basis sets up to 6-311++G(3df,2pd). The influence of SCF convergence level and density grid size on the root mean square of harmonic and anharmonic frequency deviations from experimental values was tested. The wavenumber of formaldehyde CH2 anharmonic asymmetric stretching mode is very sensitive to grid size for large basis sets; this effect is not observed for harmonic modes. BLYP-calculated anharmonic frequencies consistently underestimate observed wavenumbers. On the basis of formaldehyde anharmonic frequencies, we show that increasing the Pople basis set size does not always lead to improved agreement between anharmonic frequencies and experimental values.

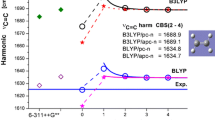
Sensitivity of water B3LYP calculated harmonic and anharmonic v s(OH) frequencies on selected Pople and polarization consistent basis sets size. The results for pc-n basis sets were fitted with two parameter formula and the CBS(2,3,4) estimated
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Apart from nuclear magnetic resonance (NMR) techniques, infrared (IR) and Raman vibrational spectroscopies are the two analytical techniques most often used for chemical characterization of small, medium and large size chemicals and their mixtures. In addition, changes in vibrational frequencies are used to study strong and weak inter- and intramolecular interactions (hydrogen bonds, association and aromatic stacking) and chemical reactions. Accurate knowledge of spectrum-molecular structure relationships is important in DNA and enzymatic studies, as well as in biochemistry and pharmacology. It is therefore obvious that theoretical predictions should provide reliable frequencies and band intensities in order to support analysis of observed vibrational spectra.
Vibrational frequencies (wavenumbers) predicted theoretically at self-consistent field (SCF), density functional theory (DFT) and second order Møller–Plesset (MP2) levels of calculations are overestimated due to anharmonicity effects [1]. This effect is most severe (over 10%) in the case of SCF predicted C–H, N–H and O–H stretching vibrations. To date, almost 4,000 papers have cited the first study in which a simple remedy was proposed to cure the deficiency in this theory by the use of scaling factors [2]. Thus, scaled theoretical wavenumbers [2–4] are used to reliably compare predicted IR and Raman spectral numbers with experimental data (we will not discuss scaling of individual force constants here). The uncertainties of combinations of 40 methods and basis sets have been studied [5]. Estimation of empirical scaling factors from analysis of numerous compounds and their fundamental vibrations is very tedious work [2]. Obviously, there are still some inherent errors in the proposed scaling factors. For example, Hartree-Fock (HF)-scaled frequencies show less uncertainty than the corresponding MP2 frequencies [5, 6]. The most often used approach is based on a single scaling factor, while more sophisticated studies use individual scaling of low and high frequencies, as well as scaling for individual modes [e.g., ν(C = O), ν(OH), ν(CH)].
Structural and vibrational parameters predicted by theoretical methods depend on the level of theory, inclusion of correlation effects, and the completeness of the one-electron basis set used. For practical reasons, DFT [7–9] including some degree of electron correlation is the best compromise between accuracy and size of the molecular system studied, and B3LYP is a typical choice of density functional.
Among the high number of basis sets available, the so-called Pople sets, though fairly old, are robust and relatively small. Sometimes they reproduce experimental parameters very well. However, there is no regular change in energy toward the complete basis set limit (CBS) calculated using Pople basis sets. Dunning and coworkers [10–13] utilized the idea of smooth and regular converging energy toward the CBS for constructing correlation-consistent basis set hierarchies [(aug)-cc-pVXZ, where X = D, T, Q, 5 and 6]. Thus, the CBS energy, and some other structural and spectral parameters were estimated using simple 2- and 3-parameter formulas. Obviously, the most accurate results were obtained for larger X (Q, 5 and 6). Later, Jensen [14–19], and also Jorge [20] designed other families of converging basis sets. In particular, Jensen’s polarized-consistent basis sets pc-n, where n = 0, 1, 2, 3 and 4 seem to converge faster than with Dunning’s sets, while reproducing the calculated parameters in the SCF, DFT, MP2 and coupled cluster, singles and doubles with triples treated approximately [CCSD(T)] basis set limits [21, 22].
Several benchmark studies have been published recently on coupled cluster (CC) predicted geometry and vibrational frequencies of selected small molecules using the correlation-consistent basis sets [23–25]. In fact, the frequencies of water [25, 26] and formaldehyde [25] have been very well reproduced using high level calculations. Unfortunately, CC methodology is prohibitively expensive for larger molecules. However, the new, less popular but more affordable pc-n basis sets were not employed in these benchmark tests. Besides, there is an open question about Kohn-Sham limiting values of vibrational frequencies obtained using harmonic and anharmonic models.
In this study we will address the problem of the accuracy of calculated harmonic and anharmonic vibrational frequencies for water and formamide in the gas phase using Pople vs Jensen’s and Dunning’s basis sets, and the convergence of individual results toward B3LYP CBS. In addition, the accuracy of the density grid in calculated harmonic and anharmonic frequencies will be tested. Water and formaldehyde were selected as simple model molecules for our study as their harmonic and anharmonic frequencies in the gas phase are well known. Several works comparing the theoretical and experimental vibrational spectra of these molecules have been published [25–29]. Moreover, their structural and vibrational parameters are modified by intermolecular interactions, including solute–solvent interactions. Thus, the conclusions of current study will aid further detailed studies on amides and small polypeptides in the gas phase and solution.
Therefore, in this work, we will test the performance of a typical, easy to compute harmonic model, and a more computationally demanding anharmonic method. Both methods are available in Gaussian 09 [30] and other software packages. We will also apply an empirical (single or global) scaling factor to harmonic frequencies and compare the results obtained with experimental and previously reported wavenumbers.
Theoretical calculations
All calculations were performed using the Gaussian 09 program [30] and some results were confirmed using Gaussian 03 [31].
Basis sets and density functionals
Pople’s 3-21G, 6-31G, 6-31G*, 6-311++G** and 6-311++G(3df,2pd), Jensen’s pc-n polarized-consistent, and Dunning’s (aug)-cc-pVXZ basis sets were used. The efficient B3LYP density functional was selected and, for comparison purposes, some calculations were also performed at restricted HF (RHF) and MP2 levels. In addition, several other common DFT methods were selected (BLYP, B3PW91 and PBE). The pc-n basis sets were downloaded from EMSL [32].
Geometry
Fully optimized geometries of water and formaldehyde in the gas phase were obtained using default and very tight convergence criteria for each method and basis set selected. All positive harmonic vibration frequencies were obtained ensuring ground state structures.
Harmonic and anharmonic vibration calculations
The calculations were carried out in the gas phase (vacuum) using the VPT2 method as implemented by Barone [33, 34] in the Gaussian program package. In several cases, the finest DFT integration grid was selected by using in the command line SCF=tight and Int(Grid=150590) instead of Int(Grid=ULTRAFINE) keyword. The use of such a fine grid is critical in the case of indirect spin–spin coupling constant calculations with tailored basis sets [35, 36].
CBS calculations
The harmonic and anharmonic frequencies, Y(x), were calculated using polarization-consistent pc-n basis sets, where n = 0, 1, 2, 3 and 4, and the correlation-consistent (aug)-cc-pVXZ basis sets, where X = D, T, Q, 5 and 6, and subsequently extrapolated to the B3LYP CBS limit, Y(∞), by fitting the results to two-parameter functions [37]:
The extrapolated value Y(∞) corresponds to the best estimate of the predicted property for infinite zeta (or cardinal number “X”), where A and Y(∞) are fitted parameters. In the case of Jensen’s pc-n basis sets, X = n + 2 was assumed for graphical fitting purposes only [21, 38]. All fittings were performed with a two-parameter formula (Eq. 1), in several cases enabling exact fitting of only two data points. Since smaller values of “X” and “n” yield results (frequencies in this study) that are more corrupted by errors due to basis set imperfections, the CBS values are often estimated using higher cardinal numbers. For example, CBS(4,5,6) indicates estimation using X = Q, 5 and 6, or n = 2, 3 and 4, respectively.
Scaling factors
Single scaling factors were used for low and high frequencies. Three fundamental studies [2–4] on scaling factors are used in frequency and zero-point vibrational (ZPV) energy calculations. Evaluation of scaling factors is very laborious work and, therefore, despite the presence of myriad methods and basis sets, only a few scaling factors are available in the literature. In particular, scaling of results obtained with the recently introduced Jensen’s basis sets and very large Dunning’s basis sets is lacking. Thus, in several cases we arbitrary used values taken from similar basis sets. For the convenience of the reader, all the scaling factors used in our work are collected in one table (Table S1 in the electronic supplementary material).
Results and discussion
The B3LYP-calculated harmonic and anharmonic frequencies of water modes as a function of selected Pople and Jensen basis set size are shown in Fig. 1. For δ(HOH) mode, the wavenumbers predicted with Pople basis sets behave irregularly, and an increase in the basis set size (compare 6-31G and 6-31G*) does not lead to better prediction of this water vibration. On the other hand, the results obtained with Jensen basis sets change more regularly. Thus, we used Eq. 1 to fit the results of both harmonic and anharmonic frequencies for n = 2, 3 and 4 toward the basis set limit. The limiting values [CBS(harm) and CBS(anharm)] are shown in Fig. 1 as straight dashed lines and compared with experimentally observed results in the gas phase (straight solid line). Usually, we observed a significantly lower sensitivity of wavenumber to the size and completeness of pc-n basis set hierarchy than with the Pople basis sets. Moreover, one can see a significantly smaller deviation from experimental values for the estimated CBS anharmonic with respect to harmonic frequencies. For example, for the water OH asymmetric stretch mode these values are −34 vs 143 cm−1, respectively (Fig. 1). B3LYP-predicted formaldehyde vibrational modes show a similar dependence on basis set type and size (Fig. 2).

Sensitivity of water B3LYP-calculated harmonic and anharmonic frequencies on selected Pople and polarization consistent basis sets size. The results for pc-n basis sets were fitted with Eq. 1 and the complete basis set limit (CBS) (2,3,4) estimated

Sensitivity of formaldehyde B3LYP-calculated harmonic and anharmonic frequencies on selected Pople and polarization consistent basis sets size. The results for pc-n basis sets were fitted with Eq. 1 and the CBS(2,3,4) estimated
One might expect that, in the case of numerical calculations of anharmonic frequencies, the quality of the results could be influenced by the accuracy of the density grid, as in the case of the indirect spin–spin coupling constant [36, 39]. Detailed analysis of water and formaldehyde B3LYP frequency deviation from experimental values [40, 41], calculated with Pople and polarization-consistent basis sets, is shown in Tables 1 and 2, respectively. Both harmonic and anharmonic deviations of water individual stretching and deformation modes are compared with deviations from simple scaling of harmonic values for different basis sets. In addition, as some general measure of calculation accuracy, the standard root mean square (RMS) deviation values are shown. The top of Table 1 gathers the results obtained for default optimization and frequency conditions (keywords OPT, Freq=anharm), and compare them with results calculated using a very accurate density grid [keywords OPT=tight, Freq=anharm, SCF=tight, INT(GRID=150590)]. Thus, the upper half of Table 1 lists results for selected Pople basis sets, and the bottom half the corresponding values obtained with Jensen’s basis sets and the final CBS values. Similar results obtained for formaldehyde are presented in the same way in Table 2. First, it is evident from Table 1 that there is no impact of grid size on the accuracy of water frequency prediction for either Pople or Jensen’s basis sets. However, in the case of high frequency formaldehyde anharmonic vibrations [νasym (CH2) in Table 2], grid size has a significant impact on the two largest Pople [6-311++G** and 6-311++G(3df,2pd)] and Jensen’s basis sets (n = 1, 2, 3 and 4 as well as CBS). Thus, a more accurate density grid is important for improving formaldehyde anharmonic frequency accuracy. On the contrary, formaldehyde harmonic frequencies do not change upon changing grid size.
There is no clear dependence of Pople basis set size on RMS deviations of harmonic and anharmonic frequencies. For example, the 6-31G basis set predicts water harmonic frequencies relatively well compared to anharmonic ones. In contrast, the same basis set (6-31G) gives the opposite result in the case of formaldehyde. Thus, we should treat such behavior as the result of accidental error cancellation. In other words, vibrational analysis using small basis sets is unreliable due to basis set incompleteness. Larger Pople basis sets are associated with an improvement in prediction of water anharmonic frequencies. Thus, for the 6-311++G(3df,2pd) basis set, corresponding anharmonic and harmonic RMS deviations of 17 vs 139 cm−1 are observed. This is also clearly visible in Fig. 1. In the case of Jensen’s basis set, starting from n = 2, water anharmonic frequencies are predicted significantly better than harmonic frequencies (RMS deviations of 23 vs 129 cm−1 for pc-2). Moreover, the RMS values for anharmonic water frequencies predicted with Pople basis sets [other than 6-311++G(3df,2pd)] are larger than with the pc-n basis set.
The use of simple harmonic frequency scaling leads to fairly accurate water wavenumbers. The accuracy of scaled water wavenumbers is similar to the anharmonic results for the studied Pople and Jensen’s basis sets (Table 1), and, for formaldehyde, scaled harmonic frequencies are often even closer to experimental values than the anharmonic frequencies (Table 2).
Next, water and formaldehyde harmonic and anharmonic wavenumbers were calculated with Dunning’s cc-pVXZ and aug-cc-pVXZ basis sets. The results were very similar to those obtained earlier with Jensen’s basis sets (see Figs. S1–S4 in the electronic supplementary material), and the corresponding deviations from experimental values are listed in Tables S2 and S3. Similarly to the results in Table 1, there is no dependence on grid size of water frequencies predicted with both Dunning’s basis set series (Table S2). However, in the case of formaldehyde, similarly to results obtained with Jensen’s basis set family (Table 2), the improvement in grid size used in conjunction with larger Dunning’s basis sets (cc-pVXZ for X = 5 and 6, and aug-cc-pVXZ for X = T, Q and 5) leads to an improvement in RMS of anharmonic frequencies of more than twofold, due mainly to a better description of CH2 asymmetric stretching. Moreover, in all cases the scaled harmonic frequencies for formaldehyde are significantly closer to experimental values than the corresponding anharmonic values (Table S3), and are comparable for water (Table S2).
The CBS values obtained with Jensen’s and Dunning’s basis set families are very similar for both molecules. However, it is important to note that Jensen’s basis sets allow significantly faster calculations than Dunning basis sets. The dependence of CPU time necessary for VPT2 calculations with pc-n, cc-pVXZ and aug-cc-pVXZ basis sets in the case of formaldehyde is presented in Fig. 3. For example, the CPU time for formaldehyde anharmonic calculations using cc-pV6Z and pc-4 basis sets with the same computer resources and configuration was 16 vs 2.5 days, respectively. Similar patterns of CPU timing are observed for water (Fig. S5). In addition, the advantage of using polarization- instead of correlation-consistent basis sets becomes more important for larger molecules.

CPU time (min) dependence on the type and size of basis set for formaldehyde VPT2 calculation with pc-n, cc-pVXZ and aug-cc-pVXZ basis sets
In the next step we tested the performance of several methods (RHF, MP2, B3LYP, BLYP, B3PW91 and PBE) in predicting anharmonic frequencies of water and formaldehyde at different Jensen’s basis set sizes (pc-2 and pc-4) and compared the results with those from two often used Pople’s basis sets (6-31G and 6-311++G**). The results obtained for water harmonic and anharmonic frequency deviations from experiment are shown in Table 3; similar data for formaldehyde are shown in Table 4. Contrary to formaldehyde anharmonic results obtained from B3LYP calculations discussed earlier, there was no influence of grid size on water and formaldehyde anharmonic deviations at BLYP, B3PW91 and PBE level. Therefore, only results for large grids and tight SCF convergence criteria are presented in Tables 3 and 4. However, for the sake of comparison, all results are presented in Tables S4–S7.
In the case of RHF calculations, both harmonic and anharmonic (although these are considerably better) frequencies obtained with both Pople and Jensen’s basis sets significantly overestimate experimental water and formaldehyde frequencies. The MP2 anharmonic values obtained with the 6-31G basis set for water and formaldehyde are not very accurate, but increasing the size of the basis set significantly improves the results. On the other hand, MP2 calculations are extremely expensive and feasible for very small molecules only. Water harmonic values obtained at the BLYP/6-31G level underestimate experimental frequencies, and anharmonic calculation using the PT2 method leads to their severe underestimation. Accidental error cancellation leads to very accurate BLYP calculated water harmonic frequencies but the corresponding anharmonic values are too low (Table 3). In the case of formaldehyde, harmonic frequencies calculated at BLYP level using larger basis sets are fairly accurate, while the corresponding anharmonic values are too small. Hence, paradoxically, formaldehyde anharmonic vibrations calculated at the BLYP level with larger basis sets exhibit worse RMS values. In the case of B3PW91 and PBE density functionals, similar improvements to those observed for B3LYP are obtained in case of formaldehyde anharmonic frequencies for larger basis sets (Tables 3, 4). However, it should be noted that, contrary to B3LYP, very good anharmonic results are obtained for formaldehyde by using the default grid size with B3PW91 and PBE density functionals (see Tables S6, S7). This makes B3LYP a more expensive DFT method for anharmonic calculations of some molecules. Therefore, to gain a more general insight, similar studies on the accuracy and reliability of the VPT2 method in predicting fundamental vibrations for a larger set of model molecules are planned.
Conclusions
In this paper we show, for the first time, the convergence of harmonic and anharmonic (calculated using VPT2 method) water and formaldehyde frequencies toward the B3LYP/pc-n and B3LYP/(aug)-cc-pVXZ CBS.
-
1.
The convergence of harmonic and anharmonic frequencies with respect to basis set size shows that pc-n basis sets consistently perform better than Pople basis sets. Both correlation-consistent and polarization-consistent basis sets enable essentially the same CBS values of harmonic and anharmonic frequencies to be obtained. However, the CPU time for calculations using cc-pVXZ basis sets is significantly longer than with the corresponding pc-n sets. The deviations in CBS values for harmonic frequencies are significantly larger than the corresponding anharmonic numbers (RMS of 119 vs 24 cm−1 in the case of water frequencies calculated using B3LYP/pc-n, and 62 vs 32 cm−1 in the case of formaldehyde frequencies). However, RMS deviations after simple scaling of harmonic frequencies are in most cases smaller and easier to obtain (39 and 16 cm−1, for water and formaldehyde, respectively). On the other hand, there are as yet no available scaling factors for Jensen’s basis set. Thus, arbitrary scaling factors were used for harmonic frequencies calculated with polarization-consistent basis sets.
-
2.
There is no point in using the VPT2 method in conjunction with the RHF and BLYP methods (the former values are far too high, and for the second method the anharmonic frequencies are too low).
-
3.
Optimization criteria and density grid size have a negligible effect on the harmonic frequencies of water and formaldehyde, but could significantly influence the corresponding anharmonic vibrations. For example, in more demanding calculations (OPT=very tight, SCF=tight and INT(GRID = 150590), the B3LYP-calculated formaldehyde anharmonic frequencies with large basis sets are significantly closer to experimental values.
The anharmonic frequencies depend on many points on the potential energy surface (PES) away from the equilibrium, and the method of calculation applied should produce very smooth PES (with constant errors). This could explain the high sensitivity of formaldehyde anharmonic frequencies to grid size, in contrast to harmonic vibrations. With the default grid size (sparse points), energy variations are not smooth and could lead to significant changes in anharmonic frequencies. On the basis of the results obtained here, we would stress the need for further study in this field.
References
Hehre WJ, Radom L, Schleyer PR, Pople JA (1986) Ab Initio molecular orbital theory. Wiley, New York
Scott AP, Radom L (1996) Harmonic vibrational frequencies: an evaluation of Hartree-Fock, Möller-Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors. J Phys Chem 100:16502–16513
Merrick JP, Moran D, Radom L (2007) An evaluation of harmonic vibrational frequency scale factors. J Phys Chem A 111:11683–11700
Sinha P, Boesch SE, Gu C, Wheeler RA, Wilson AK (2004) Harmonic vibrational frequencies: scaling factors for HF, B3LYP, and MP2 methods in combination with correlation consistent basis sets. J Phys Chem A 108:9213–9217
Irikura KK, Johnson RD III, Kacker RN (2005) Uncertainties in scaling factors for ab initio vibrational frequencies. J Phys Chem A 109:8430–8437
Dunn ME, Evans TM, Kirschner KN, Shields GC (2006) Prediction of accurate anharmonic experimental vibrational frequencies for water clusters, (H2O)n, n = 2–5. J Phys Chem A 110:303–309
Foresman JB, Frisch A (1996) Exploring chemistry with electronic structure methods. Gaussian, Pittsburg
Labanowski JK, Anzelm JW (1991) Density functional methods in chemistry. Springer, New York
Barone V (1995) In: Chong DP (ed) Recent advances in density functional methods. World Scientific, Singapore
Dunning TH Jr (1989) Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J Chem Phys 90:1007–1023
Dunning TH Jr (2000) A road map for the calculation of molecular binding energies. J Phys Chem A 104:9062–9080
Wilson A, van Mourik T, Dunning TH Jr (1996) Gaussian basis sets for use in correlated molecular calculations. VI. Sextuple zeta correlation consistent basis sets for boron through neon. J Mol Struct Theochem 388:339–349
Kendall RA, Dunning TH Jr, Harrison RJ (1992) Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J Chem Phys 96:6796–9806
Jensen F (1999) The basis set convergence of the Hartree-Fock energy for H2. J Chem Phys 110:6601–6605
Jensen F (2001) Polarization consistent basis sets: principles. J Chem Phys 115:9113–9125
Jensen F (2002) Polarization consistent basis sets: II. Estimating the Kohn-Sham basis set limit. J Chem Phys 116:7372–7379
Jensen F (2003) Polarization consistent basis sets. IV. The basis set convergence of equilibrium geometries, harmonic vibrational frequencies, and intensities. J Chem Phys 118:2459–2463
Jensen F, Helgaker T (2004) Polarization consistent basis sets. V. The elements Si–Cl. J Chem Phys 121:3463–3470
Jensen F (2005) The effect of different density functional methods on basis set parameters. Chem Phys Lett 402:510–513
Jorge FE, Sagrillo PS, de Oliveira AR (2006) Gaussian basis sets of 5 zeta valence quality for correlated wave functions. Chem Phys Lett 432:558–563
Kupka T, Lim C (2007) Polarization-consistent vs correlation-consistent basis sets in predicting molecular and spectroscopic properties. J Phys Chem A 111:1927–1932
Shahbazian S, Zahedi M (2005) Towards a complete basis set limit of Hartree-Fock method: correlation-consistent versus polarized-consistent basis sets. Theor Chem Acc 113:152–160
Tew DP, Klopper W, Heckert M, Gauss J (2007) Basis set limit CCSD(T) harmonic vibrational frequencies. J Phys Chem A 111:11242–11248
Rauhut G, Knizia G, Werner HJ (2009) Accurate calculation of vibrational frequencies using explicitly correlated coupled-cluster theory. J Chem Phys 130:054105–054110
Martin JML (1994) On the performance of correlation consistent basis sets for the calculation of total atomization energies, geometries, and harmonic frequencies. J Chem Phys 100:8186–8193
Feller D, Peterson KA (2009) High level coupled cluster determination of the structure, frequencies, and heat of formation of water. J Chem Phys 131:154306–154310
Begue D, Carbonniere P, Barone V, Pouchan C (2005) Performance of ab initio and DFT PCM methods in calculating vibrational spectra in solution: Formaldehyde in acetonitrile as a test case. Chem Phys Lett 416:206–211
Begue D, Pouchan C (2007) Vibrational anharmonic calculations in solution: performance of various DFT approaches. J Comput Chem 28:1456–1462
Daněček P, Bouř P (2007) Comparison of the numerical stability of methods for anharmonic calculations of vibrational molecular energies. J Comput Chem 28:1617–1624
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ (2009) Gaussian 09, Revision A.02. Gaussian, Wallingford
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian03, Revision E01. Gaussian, Wallingford
EMSL basis set exchange. https://bse.pnl.gov/bse/portal
Barone V (2004) Vibrational zero-point energies and thermodynamic functions beyond the harmonic approximation. J Chem Phys 120:3059–3065
Barone V (2005) Anharmonic vibrational properties by a fully automated second-order perturbative approach. J Chem Phys 122:014108–014110
Kupka T (2008) From correlation-consistent to polarization-consistent basis sets estimation of NMR spin-spin coupling constant in the B3LYP Kohn-Sham basis set limit. Chem Phys Lett 461:33–37
Krivdin LB, Sauer SPA, Peralta JE, Contreras RH (2002) Non-empirical calculations of NMR indirect carbon-carbon coupling constants: 1. Three-membered rings. Magn Reson Chem 40:187–194
Helgaker T, Klopper W, Koch H, Noga J (1997) Basis-set convergence of correlated calculations on water. J Chem Phys 106:9639–9646
Kupka T, Stachów M, Nieradka M, Kaminsky J, Pluta T (2010) Convergence of nuclear magnetic shieldings in the Kohn-Sham limit for several small molecules. J Chem Theor Comput 6:1580–1589
Kupka T (2009) Prediction of water’s isotropic nuclear shieldings and indirect nuclear spin-spin coupling constants (SSCCs) using correlation-consistent and polarization-consistent basis sets in the Kohn-Sham basis set limit. Magn Reson Chem 47:210–221
Benedict WC, Gailar N, Plyler EK (1956) Rotation-vibration spectra of deuterated water vapor. J Chem Phys 24:1139–1165
Nakanaga T, Kondo S, Saeki S (1982) Infrared band intensities of formaldehyde and formaldehyde-d2. J Chem Phys 76:3860–3865
Acknowledgments
Aneta Buczek is a recipient of a PhD fellowship from a project funded by the European Social Fund. Calculations were carried out in Wroclaw Centre for Networking and Supercomputing (http://www.wcss.wroc.pl), and in the Academic Computer Centre CYFRONET, AGH, Kraków, grant MEiN/SGI3700/UOpolski/063/2006. T. K. was supported by grant 10/WCH/2010-S.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
894_2010_913_MOESM1_ESM.doc
Additional figures (convergence of water and formaldehyde harmonic and anharmonic frequencies as well as CPU timing for water) and tables (applied scaling factors and the influence of grid size and SCF convergence criteria on harmonic and anharmonic frequency deviations) are gathered in supporting material. This material is made available free of charge via the Internet at http://pubs.acs.org. (DOC 482 kb)
Figure S1
Convergence of water harmonic and anharmonic frequencies calculated at B3LYP/cc-pVXZ level. The CBS (Q-6) values estimated using Eq. 1. (DOC 162 kb)
Figure S2
Convergence of water harmonic and anharmonic frequencies calculated at B3LYP/aug-cc-pVXZ level. The CBS (Q-6) estimated using Eq. 1. (DOC 162 kb)
Figure S3
Convergence of formaldehyde harmonic and anharmonic frequencies calculated at B3LYP/cc-pVXZ level. The CBS (Q-6) estimated using Eq. 1. (DOC 323 kb)
Figure S4
Convergence of formaldehyde harmonic and anharmonic frequencies calculated at B3LYP/aug-cc-pVXZ level. The CBS (Q,5) estimated using Eq. 1. (DOC 427 kb)
Figure S5
CPU time (min) dependence on the type and size of basis set for water VPT2 calculation with pc-n, cc-pVXZ and aug-cc-pVXZ basis sets. (DOC 88 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Buczek, A., Kupka, T. & Broda, M.A. Extrapolation of water and formaldehyde harmonic and anharmonic frequencies to the B3LYP/CBS limit using polarization consistent basis sets. J Mol Model 17, 2029–2040 (2011). https://doi.org/10.1007/s00894-010-0913-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-010-0913-3