
Abstract
A monoclonal antibody (mAb) was produced against a fluvoxamine (FLV)-bovine serum albumin conjugate that was specific to both the conjugate and free form of FLV. The mAb enabled us to develop an immunohistochemistry (IHC) method for pharmacokinetic analysis of FLV at the cell and tissue levels. We demonstrated that IHC can be used to detect the localization of FLV in the small intestine, kidney, and liver 1 h after drug administration at the cell and tissue levels. Protease digestion is an important factor for obtaining appropriate IHC staining results for localization of drugs. In this study, precise FLV localization could be determined with only 1 h of protease digestion in the kidneys, but in the small intestine and liver, the staining results with two digestive conditions had to be merged. IHC provided new findings, such as (1) nerve cells are likely to uptake more FLV than other cells and tissues; (2) the ability of reabsorption and secretion in the kidney varies depending on the site, and the amount of FLV in the primary urine is regulated downstream of the proximal tubule S3 segment; and (3) some of the FLV is excreted in the bile.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Globally, the number of individuals with depression has increased in recent years. The World Health Organization estimated the prevalence of depression as 322 million individuals worldwide in 2015 [1]. Moreover, the prevalence of depression in Japan is approximately 5.06 million individuals, which is approximately 4% of the total Japanese population, and the lifetime prevalence is even higher, reaching 6.1% [2]. Furthermore, the number of suicides worldwide in 2015 was estimated to be approximately 788,000, and the main cause of this is thought to be depression [1]. Therefore, depression has become a major social problem, both in Japan and worldwide.
In the early 1950s, the anti-tuberculosis drug isoniazid was found to induce euphoria and was used to treat depression. This marked the beginning of drug therapy for depression. However, early generation drugs have many serious side effects; thus, drugs with fewer side effects have been developed. From the end of the twentieth century, serotonin selective reuptake inhibitors (SSRI), such as fluvoxamine (FLV) and serotonin noradrenaline reuptake inhibitors (SNRI), such as milnacipran, were developed. The use of these drugs is increasing because they are considered to have fewer side effects than those by conventional antidepressants. However, they still have serious side effects, such as delirium, confusion, hallucinations, delusions, and other neuropsychiatric disorders and contribute to liver dysfunction. In addition, these drugs have been linked to an increased risk of suicidal ideations and attempts in patients aged < 24 years [3].
In recent years, “drug repositioning” or “drug repurposing” in which existing drugs are re-evaluated and diverted to treat other diseases, has become popular. An example is FLV, which is known to have anti-inflammatory effects mediated by the sigma-1 receptor and heme oxygenase-1, and serotonin reuptake inhibitory activity [4, 5]. Therefore, this drug may be used as a therapeutic agent for suppressing an excessive immune response, which can be considered as a potential risk of severeness of coronavirus disease 2019 (COVID-19) [6,7,8]. In addition, the results of several clinical trials have shown that FLV reduces the risk of COVID-19 aggravation [9, 10]. For these reasons, FLV was added to the US National Institutes of Health COVID-19 Guidelines Panel on April 23, 2021, although evidence for the efficacy of FLV was insufficient.
In vivo pharmacokinetics affect the efficacy and side effects of drugs. Pharmacokinetic data of drugs help in determining the appropriate usage. Furthermore, in drug repositioning, detailed pharmacokinetic data are useful for predicting and evaluating its efficacy and side effects and for understanding the mechanism of action. In general, in vivo pharmacokinetic studies determine drug concentrations in plasma and the resulting data are used for therapeutic drug monitoring. However, this data alone may not be sufficient to boost our understanding of the mechanism of drug action, side effects, and interactions; therefore, pharmacokinetic data in cells and tissues are also required. In this study, we report the preparation and characterization of a specific monoclonal antibody (mAb) against FLV and the development of an immunohistochemistry (IHC) method for the pharmacokinetic analysis of FLV at the cell and tissue levels.
Materials and methods
Chemicals
Fluvoxamine maleate was purchased from Nichi-Iko Pharmaceutical Co. Ltd. (Toyama, Japan). N-(γ-maleimidobutyryloxy) succinimide (GMBS) and bovine serum albumin (BSA) were obtained from the FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Protease (type XXIV: bacterial) and sodium borohydride were purchased from Sigma Aldrich Co. Inc. (St. Louis, MO, USA). Histofine® Simple Stain Rat MAX PO (M) and 3,3-diaminobenzidine tetrahydrochloride (DAB) were purchased from Nichirei Biosciences Inc. (Tokyo, Japan) and Dojindo Laboratories (Kumamoto, Japan), respectively.
Immunogen preparation (FLV-GMBS-BSA conjugate)
The immunogen was prepared according to our previous method for anti-daunomycin serum using a heterobifunctional agent GMBS [11, 12]. Briefly, FLV maleate (5 mg, 11.5 μmol) in 2.0 mL of 0.1 M phosphate buffer (PB, pH 7.0) and GMBS (1.6 mg, 5.7 μmol) in 0.5 mL tetrahydrofuran were mixed, constantly stirred, and incubated for 60 min at room temperature (RT), thus yielding a GMBS-acylated FLV solution. The sample was centrifuged for 10 min at 2000 rpm and the supernatant was collected. Acetylmercaptosuccinyl BSA (15 mg, approximately 0.1 μmol) was dissolved in 200 μL of 0.1 M PB and incubated with 50 μL of 0.5 M hydroxylamine (pH 7.4) for 10 min at RT to remove the acetyl group. The resulting mercaptosuccinyl BSA was diluted with 1 mL of 0.1 M PB, then immediately added to the GMBS-acylated FLV supernatant and incubated for 60 min with slow stirring. The conjugate was applied to a 2.5 × 45 cm Sephadex G-75 (Sigma Aldrich Co. Inc. St. Louis, MO, USA) column, previously equilibrated with 10 mM PB and eluted with the same buffer. The eluate, absorbance monitored at 280 nm, was collected in 3 mL fractions and the concentration of the conjugate was determined using the modified Lowry protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). The peak fractions were used for immunization.
Preparation of anti-FLV monoclonal antibody
Three 5-week-old female BALB/c mice were injected intraperitoneally (ip) with 100 µg FLV-GMBS-BSA conjugate emulsified in Freund’s complete adjuvant (Difco Laboratories, Detroit, MI, USA). Subsequently, they received two injections of 50 µg of the conjugate emulsified in Freund’s incomplete adjuvant at 2 week intervals. Following immunization, antisera were collected and antibody titers were evaluated using an enzyme-linked immunosorbent assay (ELISA), as described below. The mouse with the best immune response were selected for hybridization. A fourth i.p. booster injection was administered and the mice were sacrificed 4 days later. Spleen cells (2 × 108) from immunized mice and 3 × 107 myeloma cells (P3/NS-1) were fused with polyethylene glycol according to our previously reported method [13]. Cells suspended in hypoxanthine-aminopterin-thymidine (HAT) medium were dispensed at a density of 105 cells/well into 96-well tissue culture plates (Corning, New York, NY, USA) pre-seeded with 105 feeder cells. From 10 to 20 days post-fusion, the wells were screened for reactivity using ELISA, as described below. Limiting dilutions of positive cultures were carried out two or three times to obtain monoclonality, and sub-isotyping of the mAbs was performed using a mouse monoclonal sub-isotyping kit (American Qualex Manufactures, San Clemente, CA, USA).
Dilution ELISA (evaluation of antibody titers)
Dilution ELISA was performed as previously described for anti-alogliptin monoclonal antibodies [14]. The wells of the microtiter plates were coated with 100 μL of the FLV–GMBS–BSA conjugate (10 µg/mL) or BSA (10 µg/mL) for 60 min at 37 °C. The protein binding sites were blocked with 1% skimed milk for 30 min at RT. The wells were then incubated with serially diluted antiserum (to assess whether the serum contained antibodies to FLV conjugate) or hybridoma culture supernatant (to screen clones producing antibodies against FLV conjugate, or evaluation of monoclonal antibody titer) for 90 min at 37 °C, followed by incubation with Simple Stain™ Rat MAX PO (M) (1:1000; 100 µL) for 60 min at 37 °C. The amount of enzyme conjugate bound to each well was measured using O-phenylenediamine (OPD; FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) as a substrate, and the absorbance at 492 nm was measured using an automatic ELISA analyzer (ImmunoMini NJ-2300; Nalje Nunc Int. Co., Ltd., Tokyo, Japan).
Inhibition ELISA (evaluation of antibody specificity)
Inhibition ELISA was performed to evaluate the specificity of mAb according to our previous method [14]. Wells in a microtiter plate were coated with FLV–GMBS–BSA conjugate and blocked with skimmed milk. A fixed concentration of anti-FLV antibody (1:2,000; 50 µL) and different concentrations of FLV–GMBS–BSA, FLV, or FLV acid (the main metabolite of FLV) (50 µL) were added to the wells. The plates were then incubated for 90 min at 37 °C. After adding Simple Stain™ Rat MAX PO (M) (1:1000; 100 µL) and incubating the plates for 60 min at 37 °C, the bound enzyme activity was measured as described above.
Binding ELISA (evaluation of antibody specificity and simulation of IHC)
Binding ELISA was performed to evaluate the specificity of the mAb and determine whether it could be used in IHC. This method is a simulation of IHC because it binds the drug to coated amino acids in situ using a cross-linking agent, glutaraldehyde (GA). Following our previous method for vancomycin (VCM) [15], wells in a microtiter plate coated with poly-l-lysine (30 µg/mL) were activated with 2.5% GA in 50 mM borate buffer (pH 10.0) for 1 h. The plates were then incubated with different concentrations of the test compounds for 1 h at RT, after which excess aldehyde groups were blocked with 1% sodium borohydride. The plates were further incubated for 1 h with 1% skimmed milk and then overnight at 4 °C with the primary antibody anti-FLV mAb diluted 1:100 in phosphate-buffered saline (PBS; 10 mM phosphate buffer, pH 7.2, containing 0.15 M NaCl) containing 0.05% Tween 20. Subsequently, incubation with the Simple Stain™ Rat MAX PO (M) (1:1000; 100 µL) and measurement of the bound enzyme activity were performed in the same manner as described for dilution ELISA.
Animals
Male adult Wistar rats (CLEA Japan, Inc., Tokyo, Japan), 200–250 g in body weight, were used in this study. All maintenance and experiments were conducted in accordance with the principles of laboratory animal care and national laws. The animals were housed in temperature- and light-controlled rooms (21 ± 1 °C, 12 h light:12 h dark) with free access to food and tap water. Three rats were orally administered a single dose of 10 mg FLV/kg body weight. After 1 h, the rats were anesthetized with sodium pentobarbital (60 mg/kg; Abbott Laboratories, North Chicago, IL, USA) and perfused transcardially with PBS containing 5,000 IU of heparin sodium (AY Pharmaceuticals Co., Ltd. Tokyo, Japan) at 50 mL/min for 2 min at RT and then with a freshly prepared solution of 2% GA in PBS for 6 min. The small intestine (duodenum), kidneys, and liver were quickly excised and fixed in the same fixative for 5 h at RT.
Immunohistochemistry
The IHC method was essentially the same as that described in our previous studies [16, 17]. Briefly, the fixed specimens were embedded in paraffin according to a routine method, and 5 μm-thick sections were prepared. The sections were deparaffinized, rehydrated, and then consecutively treated with 6% hydrogen peroxide (H2O2) in PBS for 30 min, 2 N HCl for 30 min, no protease digestion or 0.03 mg/mL protease in PBS for 1 or 2 h at 30 °C [18], and 5 mg/mL sodium borohydride in PBS for 10 min [19, 20]. After each process, the specimens were washed three times with PBS. Next, the specimens were exposed to a blocking solution containing 10% normal goat serum (NGS), 1.0% BSA and 0.1% saponin in Tris-buffered saline (TBS; 50 mM Tris buffer, pH 7.4, containing 0.15 M NaCl) for 1 h at RT and then directly incubated with anti-FLV mAb (AFLV-65–2-2), diluted 1:20 in TBS supplemented with 1% NGS, 0.25% BSA and 0.1% saponin at 4 °C overnight. The sections were washed three times with TBS containing 0.1% Triton X-100 (TBST), twice with TBS, and then incubated with Simple Stain™ Rat MAX-PO (M) for 2 h at RT. After rinsing with TBS, the site of the antigen–antibody reaction was revealed by incubating sections in DAB (a colorimetric substrate) and H2O2 for 10 min.
Three types of negative control experiments were performed. One was an absorption control, in which an excessive amount of FLV–GMBS–BSA conjugate (30 μg/mL) was added to the antibody solution before adding the antibody to the sections. The second was a conventional control in which normal mouse IgG was incubated with the sections instead of the primary mAb. The third was immunostaining using untreated (not administered FLV) rat tissues.
Results
Generation and specificity of mAb to FLV
The ELISA screening and limiting dilution experiments were repeated and four hybridoma lines (clone no. 14-2, 42-33, 65-2-2, 65-9-1) were obtained, which secreted antibodies bound to the FLV–GMBS–BSA conjugate but did not recognize BSA. Antibodies were secreted into the culture supernatants. Hybridoma culture supernatants were used as mAbs and labeled AFLV-14-2, AFLV-42-33, AFLV-65-2-2, and AFLV-65-9-1, respectively. The sub-isotype of these mAbs was determined to be IgG1 using a mouse monoclonal sub-isotyping kit. Because the results of the experiments with these four mAbs were not very different, only the results of AFLV-65-2-2, which showed the best results, are described below. The IgG concentration of AFLV-65-2-2 was 13.39 μg/mL as determined using a mouse IgG immunoassay kit (PerkinElmer Inc., Waltham, MA, USA).
Dilution ELISA
Dilution ELISA was performed to assess antibody binding using serial dilutions of anti-FLV antibody. Serially diluted AFLV-65-2-2 mAb showed high binding activity, even at a dilution of 10,000 times (Fig. 1). In contrast, the mAb did not bind to BSA, even at low dilution rates (high concentrations of the antibody) (Fig. 1).

Evaluation of antibody titer. ELISA measurements of the binding of serially diluted anti-fluvoxamine (FLV) monoclonal antibody (AFLV-65-2-2) to the solid phase coated with FLV-GMBS-BSA (closed circles) or BSA (open circles)
Inhibition ELISA
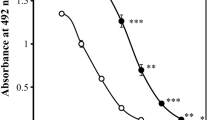
Inhibition ELISA is based on the principle that various amounts of analytes and a fixed amount of FLV conjugate compete for a limited number of binding sites on anti-FLV antibodies. A fixed amount of FLV–GMBS–BSA was coated on the wells of a microtiter plate, and anti-FLV mAb and different concentrations of analytes (FLV–GMBS–BSA, FLV, and FLV acid) were added simultaneously. Calibration curves were plotted to show the relationship between analyte concentrations and the percentage of bound antibodies. Dose-dependent inhibition curves were generated for the FLV–GMBS–BSA conjugate in the range of 15 pM to 1.5 μM and free FLV and FLV acid from 2 nM to 200 μM (Fig. 2). The doses required for 50% inhibitory concentration (IC50) using FLV–GMBS–BSA and FLV were 7 nM and 3 μM, respectively; however, no inhibition was detected with FLV acid, even at concentrations up to 20 μM (Fig. 2).

Evaluation of antibody specificity. ELISA measurements showing competition between conjugated fluvoxamine (FLV), free FLV, and FLV acid coated to the solid phase for binding to the AFLV-65-2-2. The curves show the amount (percentage) of bound enzyme activity (B) for different doses of FLV-GMBS-BSA (closed circles), FLV (closed squares), and FLV acid (open squares) as a ratio of that bound using the horseradish peroxidase-labeled second antibody alone (B0). The concentrations of FLV conjugate were calculated assuming that the molecular weight of the conjugate was 66 kDa, which is that of BSA, because the molecular weight of the drug is smaller than BSA
Binding ELISA
The binding ELISA simulates the IHC of tissue sections by coupling the amino groups of the analyte to poly-l-lysine coated on the wells of a microtiter plate using GA. The concentration of each analyte applied to the wells and the bound HRP activity was plotted to yield a dose-dependent curve with FLV concentrations ranging from 2 µM to 5 mM (Fig. 3). In contrast, no immunoreactivity with amlodipine or VCM was observed at any concentration (Fig. 3).

Evaluation of antibody specificity and simulation of immunohistochemistry. Reactivity of AFLV-65-2-2 determined from its immunoreactivity in the binding ELISA. Activated wells prepared for the binding ELISA were incubated with various concentrations of FLV (closed squares), amlodipine (open rhombuses), or vancomycin (closed rhombuses). The wells were treated with NaBH4 and then with Simple Stain Rat MAX PO (M) (1:1000; 100 µL)
Localization of FLV in rat tissues
Small intestine (duodenum)
Without protease digestion, weak to moderate staining was observed in the lamina propria mucosae and lamina submucosa, whereas the Meissner plexus and Auerbach plexus showed stronger staining intensity than them (Fig. 4a). Absorptive epithelial cells were moderately stained for brush borders; however, the cytoplasm and nuclei were almost negative (Fig. 4a, d). The intestinal crypt cells, lamina muscularis mucosae, and muscular layers were barely stained (Fig. 4a). When the sections were digested with protease for 1 h, the signals were enhanced at all sites of the sections, and in particular, the staining intensity of the muscularis mucosae and muscular layers was greatly increased (Fig. 4b). Moderate staining and weak staining of intestinal crypt cells and nuclei of absorptive epithelial cells, respectively, were also observed (Fig. 4b, e). Furthermore, prolonging protease digestion to 2 h enhanced the signals of absorptive epithelial and intestinal crypt cells (Fig. 4c, f). However, the staining intensity of the lamina propria and submucosa was almost the same as that of 1 h digested sections and that of the muscularis mucosae and muscular layer was attenuated (Fig. 4c).

Immunohistochemistry for FLV in rat small intestine (duodenum). a, d: Without protease digestion, b, e, g, h, i: 1 h protease digestion, c, f: 2 h protease digestion. i: absorption control. d, e, f: the high magnification images of the box area of a, b, c. g: Meissner’s plexus. h: Auerbach’s plexus. V: villi, C: intestinal crypt, arrow: brush border, closed arrow head: Meissner’s plexus, open arrow head: Auerbach’s plexus. Bars: a–c, i = 100 μm; d–h = 20 μm
The absorption controls showed that the addition of an excess amount of FLV conjugate to the primary antibody solution abolished all staining (Fig. 4i). Conventional control and untreated rat tissues were not stained (Supplementary figure). Moreover, no staining was observed in samples without protease digestion in all control experiments (data not shown).
Kidney
The upper layer of the kidney refers to the area where renal corpuscles are observed. In this area, we can identify the proximal tubule (segments S1 and S2) with a brush border, the distal tubules without a brush border, and collecting ducts made of two types of cells (strongly stained cells and weakly or no stained cells). The lower layer is the area where the renal corpuscles are absent and the proximal tubule S3 segment has a thick brush border. Without protease digestion, the most noticeable staining was observed in the S3 segment of the proximal tubule (Fig. 5a, e). The cytoplasm and nuclei of epithelial cells showed weak to moderate positive staining, whereas microvilli were negative (Fig. 5e). In contrast, in the S1 and S2 segments of the proximal tubule, only faint staining was observed in the microvilli of the epithelial cells, and neither the nucleus nor the cytoplasm was stained (Fig. 5a, d). In the distal tubule, a positive signal was observed in a part of the epithelial cells (Fig. 5d). Both positive and almost negative cells were found in the collecting ducts (Fig. 5d). Glomeruli were negative (Fig. 5a, d). One hour of protease treatment altered the staining pattern and intensity of the tissue. The most noticeable change was that glomeruli showed positive signals (Fig. 5b, f). Staining of the microvilli of the S1 and S2 segment cells was also enhanced, but staining intensity of the nucleus and cytoplasm was very weak (Fig. 5f). The nucleus and cytoplasm of S3 segment cells showed moderate to strong staining, but the microvilli were not stained (Fig. 5g). The staining intensity of the distal tubules and collecting ducts was also increased (Fig. 5f). Digestion of the sections with protease for 2 h did not enhance the staining at each site of the kidney (Fig. 5c, h, i). No staining was observed in any of the control experiments (Supplementary figure). The samples without protease digestion also showed no staining in all control experiments (data not shown).

Immunohistochemistry for FLV in rat kidney. a, d, e: without protease digestion, b, f, g: 1 h protease digestion, c, h, i: 2 h protease digestion. d, e: the high magnification images of the box area of a. f, g: the high magnification images of the box area of b. h, i: the high magnification images of the box area of c. G: glomerulus, S1, 2: S1 or S2 segment of proximal tubules, S3: S3 segment of proximal tubules, D: distal tubules, C: collecting ducts. Bars: a–c = 200 μm; d–i = 50 μm
Liver
Hepatocytes were not stained without protease digestion, but dot-like and linear staining was observed between the hepatocytes (Fig. 6a–c). These signals were considered indicative of bile canaliculi because of their size and location. Staining of bile canaliculi differed depending on the region of the hepatic lobule. The contents of bile canaliculi were also stained in zone III (area around the central vein) (Fig. 6b), but only the wall of canaliculi (cell membrane of hepatocytes) in zone I (peripheral area of the hepatic lobule) showed staining (Fig. 6c). Moderate staining was observed in the interlobular connective tissue (Fig. 6a, c). In the hepatic triad, only the apical side of interlobular bile duct epithelial cells showed strong staining (Fig. 6c). Endothelial cells of the hepatic sinusoid and Kupffer cells were not stained (Fig. 6b, c). After 1 h of protease digestion, the interlobular triad, interlobular connective tissue, and also the endothelial cells of sinusoid and Kupffer cells, which were not stained in the undigested sections, demonstrated strong positive signals (Fig. 6d, e, f). In addition, weak staining was observed in the nuclei and cytoplasm of hepatocytes (Fig. 6e, f). However, the signal on the bile canaliculi was attenuated; in particular, the staining of the contents of the canaliculi in zone III disappeared (Fig. 6e). Even after 2 h of protease digestion, the staining intensity at each site did not increase and the morphology degraded (data not shown). No staining was observed in any of the control experiments (Supplementary figure). The samples without protease digestion also showed no staining in all control experiments (data not shown).

Immunohistochemistry for FLV in liver. a–c: without protease digestion, d–f: 1 h protease digestion. CV: central vein, IV: interlobular vein, IA: interlobular artery, IB: interlobular bile duct, arrows: bile canaliculi, arrow heads: endothelial cells, open arrow heads: Kupffer cells. Bars: a, d = 100 μm; b, c, e, f = 25 μm
Discussion
In this study, we prepared anti-FLV mAbs, characterized their specificity, and developed an IHC procedure to determine the sites of FLV accumulation in the small intestine, kidneys, and liver of rats, which are the main organs responsible for drug absorption, metabolism, and excretion. The AFLV-65-2-2 mAb was demonstrated to be specific to FLV by inhibition and binding ELISAs. Inhibition ELISA showed that antibody binding was strongly inhibited by FLV–GMBS–BSA and FLV but not by FLV acid. The results of the binding ELISA showed that the mAb binds to FLV–GA–lysine but does not bind to amlodipine and VCM–GA–lysine. These results strongly suggest that the AFLV-65-2-2 mAb can recognize the drug with high specificity and could be useful for IHC studies of drug localization in GA-fixed tissues.
The staining intensity in IHC depends on the amount of antigen and accessibility of the antibody to the antigen. IHC for determining localization of drugs requires GA fixation to retain small-molecule drugs in situ. Therefore, pretreatments, especially protease digestion, to increase the permeability of the antibody are important to obtain precise drug localization. Enhancing protease digestion generally increases the signal intensity and reveals new positive sites. However, excessive digestion destroys the structure and the amount of antigen decreases because peptide and protein antigens are digested, and in the case of drug antigens, the amount of scaffold protein is reduced, making it difficult to retain the drug in situ. Therefore, signal in undigested or weakly digested sections may sometimes be attenuated or absent [21]. In this study, we examined how the intensity of protease digestion for each tissue sample affected the staining results and developed an IHC method for localization of FLV. The results revealed that the appropriate digestion conditions differ among epithelial cells, nerve cells, and smooth muscle cells in the small intestine. Thus, it is necessary to comprehensively determine the localization by integrating the data under multiple digestive conditions to understand the precise drug localization.
Most orally administered drugs are absorbed in the small intestine, but absorptive epithelial cells are only stained with brush borders without protease digestion. However, weak to moderate staining of connective tissue and nerve cells made it difficult to explain where the drugs in these sites came from. Through enhanced protease digestion, positive signals were detected in the nuclei and cytoplasm of absorptive epithelial cells, confirming that FLV was absorbed from this site. As mentioned above, the staining intensity of each tissue or cell is determined by the amount of antigen and antibody accessibility; therefore, nerve cells, which showed moderate staining even without protease digestion, were considered to uptake larger amounts of drug compared to those by other cells. FLV accumulated in nerve cells disturbs the function of Meissner’s and Auerbach’s plexuses, resulting in depression of intestinal secretion and motility, and may cause side effects, such as diarrhea and constipation.
In the kidney, the proximal tubule S3 segments, distal tubules, and collecting ducts showed moderate or high staining without protease digestion, but the glomeruli were negative, and the proximal tubule S1 and S2 segments showed only faint staining in the brush border of the epithelial cells. Protease digestion for 1 h enhanced the staining intensity of positive sites and made glomeruli positive; however, the staining intensity of the proximal tubule S1 and S2 segments was very weak, except for the brush border of epithelial cells. In addition, protease digestion for 2 h did not change the staining pattern and intensity of the kidney sections. Therefore, protease digestion for 1 h was considered to provide the necessary data in the kidney. Drug excretion from the kidney is determined by the relative rates of glomerular filtration, tubular reabsorption, and tubular secretion. Therefore, immunostaining of tubular epithelial cells is believed to represent the difference between drug reabsorption from primary urine and drug secretion. The results of this study suggest that the ability of reabsorption and secretion between each site of the renal tubule and collecting duct is different and that the amount of FLV in the primary urine is regulated downstream of the proximal tubule S3 segment. Although both positive and almost negative cells were found in the collecting ducts, because the collecting duct comprises two types of cells (principal and intercalated cells) [22], the difference in FLV accumulation may reflect that observed in cell types.
The hepatic lobule was divided into zone I (peripheral area of the hepatic lobule), zone II (intermediate area), and zone III (area around the central vein), according to differences in the metabolic ability of hepatocytes. Without protease digestion, the interlobular connective tissue showed moderate staining, while moderate (zone I) to strong (zone III) staining was observed in the bile canaliculi. Protease digestion for 1 h increased the staining intensity of the hepatic triad and interlobular connective tissue, while weak staining was observed in hepatocytes, but the staining of bile canaliculi was attenuated. Protease digestion for 2 h further attenuated the signal intensity in the bile canaliculi and degraded the liver morphology. Therefore, in the liver, the data without protease digestion should be presented when investigating bile canaliculi, and the data obtained with protease digestion for 1 h for other structures and cells. FLV is oxidatively demethylated in the liver to inactive metabolites (FLV acid) and is excreted in the urine. However, our results suggest that a small amount of unchanged FLV is excreted in the bile. In addition, since the staining intensity of the bile canaliculi in zone III was stronger than that in zone I, our findings suggest that the FLV uptake and excretion ability of hepatocytes differ depending on the zone, and this ability was higher in zone III than that in zone I.
Conclusion
We succeeded in producing a specific monoclonal antibody with high binding activity to FLV and in developing an IHC method that is useful for analyzing the localization of FLV using this antibody. Although appropriate experimental conditions must be determined to obtain good staining results in the IHC method, protease digestion is a particularly important factor in IHC to determine localization of drugs. In this study, precise FLV localization could be understood with only 1 h of protease digestion for the kidney, but the staining results with 1 and 2 h of digestion for the small intestine and with undigested and 1 h of digestion for the liver were merged. This study provided new insights reporting that: (1) nerve cells are likely to uptake more FLV than that by other cells and tissues; (2) the reabsorption and secretion ability in the kidney varies depending on the site, and the amount of FLV in the primary urine is regulated downstream of the proximal tubule S3 segment; and (3) some of the FLV is excreted in the bile. This study demonstrated the potential of IHC in determining the pharmacokinetics of FLV in cells and tissues. Future studies should focus on changes over time in FLV localization in these organs and should investigate FLV localization in other organs as well.
References
World Health Organization (2017) Depression and other common mental disorders. global health estimates. https://apps.who.int/iris/bitstream/handle/10665/254610/WHO-MSD-MER-2017.2-eng.pdf?sequence=1&isAllowed=y
Ishikawa H, Kawakami N, Kessler RC, World mental health Japan survey collaborators (2016) Lifetime and 12-month prevalence, severity and unmet need for treatment of common mental disorders in Japan: results from the final dataset of world mental health Japan survey. Epidemiol Psychiatr Sci 25:217–229
Nichi-Iko Pharmaceutical Co. Ltd (2021) Fluvoxamine maleate, interview form. https://www.info.pmda.go.jp/go/interview/1/530169_1179039F1168_1_015_1F.pdf
Anderson G, Maes M, Markus RP, Rodriguez M (2015) Ebola virus: melatonin as a readily available treatment option: ebola and melatonin. J Med Virol 87:537–543
Almási N, Török S, Dvorácskó S, Tömböly C, Csonka Á, Baráth Z, Murlasits Z, Valkusz Z, Pósa A, Varga C, Kupai K (2020) lessons on the sigma-1 receptor in TNBS-induced rat colitis: modulation of the UCHL-1, IL-6 pathway. Int J Mol Sci 21:4046
Hooper PL (2020) COVID-19 and heme oxygenase: novel insight into the disease and potential therapies. Cell Stress Chaperones 25:707–710
Hooper PL (2022) Heme oxygenase agonists-fluvoxamine, melatonin-are efficacious therapy for Covid-19. Cell Stress Chaprones 27:3–4
Facente SN, Reiersen AM, Lenze EJ, Boulware DR, Klausner JD (2021) Fluvoxamine for the early treatment of SARS-CoV-2 infection: a review of current evidence. Drugs 81:2081–2089
Lenze EJ, Mattar C, Zorumski CF, Stevens A, Schweiger J, Nicol GE, Miller JP, Yang L, Yingling M, Avidan MS, Reiersen AM (2020) Fluvoxamine vs placebo and clinical deterioration in outpatients with symptomatic COVID-19: a randomized clinical trial. JAMA 324:2292
Reis G, Dos Santos Moreira-Silva EA, Silva DCM, Thabane L, Milagres AC, Ferreira TS, Dos Santos CVQ, de Souza Campos VH, Nogueira AMR, de Almeida APFG, Callegari ED, de Figueiredo Neto AD, Savassi LCM, Simplicio MIC, Ribeiro LB, Oliveira R, Harari O, Forrest JI, Ruton H, Sprague S, McKay P, Glushchenko AV, Rayner CR, Lenze EJ, Reiersen AM, Guyatt GH, Mills EJ, TOGETHER Investigators (2022) Effect of early treatment with fluvoxamine on risk of emergency care and hospitalisation among patients with COVID-19: the together randomised, platform clinical trial. Lancet Glob Health 10:e42–e51
Fujiwara K, Yasuno M, Kitagawa T (1981) Novel preparation method of immunogen for hydrophobic hapten, enzyme immunoassay for daunomycin and adriamycin. J Immunol METHODS 45:195–203
Fujiwara K, Yasuno M, Kitagawa T (1981) Distribution of anticancer antibiotic daunomycin in the rat heart and kidney revealed by immunocytochemistry using monoclonal antibodies. Histochem Cell Biol 127:69–77
Fujiwara K, Shin M, Hougaard DM, Larsson L-I (2007) Distribution of anticancer antibiotic daunomycin in the rat heart and kidney revealed by immunocytochemistry using monoclonal antibodies. Histochem Cell Biol 127:69–77
Yamamoto Y, Yamamoto Y, Saita T, Shin M (2015) immunohistochemistry for anti-diabetes drug, alogliptin using a newly prepared monoclonal antibody: its precise localization in rat small intestine. Acta Histochem Cytochem 52:27–34
Fujiwara K, Yoshizaki Y, Shin M, Miyazaki T, Saita T, Nagata S (2012) immunocytochemistry for vancomycin using a monoclonal antibody that reveals accumulation of the drug in rat kidney and liver. Antimicrob Agents Chemother 56:5883–5891
Yamamoto Y, Yamamoto Y, Saita T, Hira D, Chijiwa T, Shin M (2020) Immunohistochemical pharmacokinetics of the anti-diabetes drug alogliptin in rat kidney and liver. Acta Histochem Cytochem 53:55–60
Yamamoto Y, Yamamoto Y, Saita T, Shin M (2021) Immunohistochemical localization and pharmacokinetics of the anti-MRSA drug teicoplanin in rat kidney using a newly developed specific antibody. Med Mol Morphol 54:227–236
Finley JC, Grossman GH, Dimeo P, Petrusz P (1978) Somatostatin-containing neurons in the rat brain: widespread distribution revealed by immunocytochemistry after pretreatment with pronase. Am J Anat 153:483–488
Clancy B, Cauller LJ (1998) Reduction of background autofluorescence in brain sections following immersion in sodium borohydride. J Neurosci Methods 83:97–102
Haraguchi CM, Yokota S (2002) Immunofluorescence technique for 100-nm-thick semithin sections of Epon-embedded tissues. Histochem Cell Biol 117:81–85
Fujiwara K, Shin M, Miyazaki T, Maruta Y (2011) Immunocytochemistry for amoxicillin and its use for studying uptake of the drug in the intestine, liver, and kidney of rats. Antimicrob Agents Chemother 55:62–71
Madsen KM, Tisher CC (1986) Structural-functional relationship along the distal nephron. Am J Physiol 250:F1–F15
Acknowledgements
We would like to thank Editage (http://www.editage.com) for English language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
795_2022_337_MOESM1_ESM.pdf
Supplementary file1 Supplementary figure. Three types of negative control experiments. a, d, g: absorption control. b, e, h: conventional control. c, f, i: untreated (not administered FLV) rat tissue. a, b, c: duodenum. d, e, f: kidney. g, h, i: liver. a–i: 1 h protease digestion. V: villi, C: intestinal crypt, G: glomerulus, CV: central vein, IV: interlobular vein. Bars: a-c, g-h = 100 μm; d, e, f = 20 μm (PDF 2209 KB)
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shin, M., Yamamoto, Y., Kataoka, H. et al. Development of immunohistochemistry for detecting fluvoxamine in rat tissues using newly prepared monoclonal antibody: its precise localization in small intestine, kidney, and liver of rats. Med Mol Morphol 56, 28–37 (2023). https://doi.org/10.1007/s00795-022-00337-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00795-022-00337-6