
Abstract
The complete nucleotide sequence of plasmid pP62BP1 (34,467 bp), isolated from Arctic Psychrobacter sp. DAB_AL62B, was determined and annotated. The conserved plasmid backbone is composed of several genetic modules, including a replication system (REP) with similarities to the REP region of the iteron-containing plasmid pPS10 of Pseudomonas syringae. The additional genetic load of pP62BP1 includes two highly related type II restriction-modification systems and a set of genes (slfRCHSL) encoding enzymes engaged in the metabolism of organic sulfates, plus a putative transcriptional regulator (SlfR) of the AraC family. The pP62BP1 slf locus has a compact and unique structure. It is predicted that the enzymes SlfC, SlfH, SlfS and SlfL carry out a chain of reactions leading to the transformation of alkyl sulfates into acyl-CoA, with dodecyl sulfate (SDS) as a possible starting substrate. Comparative analysis of the nucleotide sequences of pP62BP1 and other Psychrobacter spp. plasmids revealed their structural diversity. However, the presence of a few highly conserved DNA segments in pP62BP1, plasmid 1 of P. cryohalolentis K5 and pRWF-101 of Psychrobacter sp. PRwf-1 is indicative of recombinational shuffling of genetic information, and is evidence of lateral gene transfer in the Arctic environment.
Similar content being viewed by others


Introduction
In recent years, microorganisms adapted to extremely cold environments have become an area of growing scientific interest. This is mainly due to the fact that they represent a likely source of cellular products with potential biotechnological applications, such as cold-adapted enzymes (Cavicchioli et al. 2002). There is an increasing requirement for the development of efficient genetic systems that are functional at low temperatures for the expression and purification of these proteins as well as other heterologous proteins characterized by their heat lability, toxicity towards the host cell or tendency to form inclusion bodies in standard expression hosts (Werbowy et al. 2009). The development of such genetic tools is tightly linked with genomic studies on psychrophilic strains, and especially their indigenous extrachromosomal replicons. To date, a relatively small number of plasmids residing in psychrophilic strains have been characterized (e.g. Cieśliński et al. 2008). This may explain why only a few low temperature expression systems are currently available (e.g. Zhao et al. 2011; Parrilli et al. 2008; Miyake et al. 2007).
In this study, we analyzed the genetic organization of plasmid pP62BP1 of psychrophilic Psychrobacter sp. DAB_AL62B. This strain was isolated from guano sediments collected from a breeding colony of little auks (Alle alle) in the vicinity of the Polish Polar Station, situated in the Norwegian Svalbard archipelago (77°0′0″N, 15°33′0″E) in the Arctic.
Psychrophilic bacteria of the genus Psychrobacter successfully colonize diverse ecological niches in permanently cold or even permafrost regions, as well as in other low water activity environments (Rodrigues et al. 2009). Current knowledge concerning the pool of mobile DNA of Psychrobacter spp. is extremely limited. Complete nucleotide sequences of only five plasmids have been obtained to date: pRWF101 and pRWF102 of Psychrobacter sp. PRwf-1, pTAUp and pTADw of Psychrobacter sp. TA144, and plasmid 1 of P. cryohalolentis K5. The largest replicons, pRWF101 (13.9 kb) and plasmid 1 of P. cryohalolentis K5 (41.2 kb), contain 14 and 44 open reading frames (ORFs), respectively, while the other plasmids are small (1.3–2.1 kb) cryptic replicons, encoding one or two ORFs. An in-depth sequence analysis has only been reported in the case of the plasmids harbored by the strain TA144 (Tutino et al. 2000; Duilio et al. 2001).
The detailed genomic analysis of plasmid pP62BP1 of Psychrobacter sp. DAB_AL62B presented in this report includes the characterization of a variety of genetic modules that may be of great value in the construction of genetic cassettes to be used in the creation of versatile vectors functional in psychrophilic bacteria.
Materials and methods
Bacterial strain and culture conditions
The strain DAB_AL62B isolated from little auk guano collected from Spitsbergen Island in the Arctic was classified as a member of the Psychrobacter genus due to 16S rRNA sequence similarity. The strain was cultured in LB (lysogeny broth) medium (Sambrook and Russell 2001) at 21°C.
Plasmid DNA isolation
Plasmid DNA was isolated using a large-scale alkaline extraction method and purified by CsCl-ethidium bromide gradient centrifugation (Sambrook and Russell 2001).
DNA sequencing
The complete nucleotide sequence of plasmid pP62BP1 was determined in the DNA Sequencing and Oligonucleotide Synthesis Laboratory (oligo.pl) at the Institute of Biochemistry and Biophysics, Polish Academy of Sciences. High-throughput sequencing of the MID-tagged shotgun plasmid-library was performed using an FLX Titanium Genome Sequencer (Roche/454 Life Sciences). Newbler de novo assembler software (Roche) was used for the sequence assembly. Final gap closure and sequence polishing were performed by capillary sequencing of PCR products using an ABI3730xl DNA Analyzer (Applied Biosystems).
Bioinformatics
Bioinformatic characterization of the nucleotide sequence of pP62BP1 was performed using specialized software tools. The sequence was initially analyzed using ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html), Clone Manager (Sci-Ed8) and Artemis (Rutherford et al. 2000). Similarity searches were performed using the BLAST programs (Altschul et al. 1997), PRIAM (Claudel-Renard et al. 2003) and REBASE (Roberts et al. 2010). Helix-turn-helix motifs were identified by the application of GYM 2.0 (Gao et al. 1999; Narasimhan et al. 2002) and Helix-Turn-Helix Motif Prediction (Dodd and Egan 1990). Other conserved domains and motifs were identified using Motif Scan (Pagni et al. 2007), the NCBI Conserved Domain Database (CDD; Marchler-Bauer et al. 2011) and the Pfam database (Finn et al. 2010). Sequence alignments were performed using MUSCLE (Edgar 2004). Putative promoter sequences were predicted using BPROM (http://www.softberry.com/berry.html). Protein secondary structures were determined with the application of PredictProtein (Rost et al. 2003) and YASPIN (Lin et al. 2005). Protein signal sequences were identified using PrediSi (Hiller et al. 2004).
Nucleotide sequence accession number
The nucleotide sequence of plasmid pP62BP1 determined in this study has been annotated and deposited in the GenBank database with the accession number JQ065022.
Results and discussion
Overall features of pP62BP1
Plasmid pP62BP1 is the only extrachromosomal replicon residing in Psychrobacter sp. DAB_AL62B. The complete nucleotide sequence of pP62BP1 (34,467 bp) revealed the presence of 33 open reading frames (ORFs) (Fig. 1), which constitute 81.5% of the plasmid genome. In silico sequence analysis permitted the assignment of putative functions to 18 of the predicted ORFs. The remaining ORFs show the highest level of similarity (on the amino acid sequence level) to proteins of unknown function (12 ORFS), or have no relatives in the open databases (3 ORFs) (Table 1). The overall G+C content of the pP62BP1 nucleotide sequence (36.5%) is lower than the values determined for the plasmid or chromosomal DNAs of other Psychrobacter spp. strains (38.0–46.0%).

Circular representation of pP62BP1. Open reading frames (ORFs) are represented by block arrows on the outer circle. Predicted functions/homologies of the ORFs are indicated by the color key. Several ORF numbers and names are given for reference (see Table 1). The positions of the putative oriV and parS sequences are indicated by white ellipses (see text for details). Solid black blocks represent the genetic modules identified in the plasmid: REP, replication module; PAR, partitioning system; R-M1 and R-M2, restriction-modification systems; SLF, phenotypic module; MRS, multimer resolution system; TA, toxin–antitoxin system. The inner circle is a graduated scale (kb)
Comparative sequence analysis identified several putative genetic modules within the pP62BP1 genome. The plasmid backbone is composed of a replication system (REP) and three putative stabilization systems, responsible for active partitioning (PAR), resolution of multimeric plasmid forms (MRS) and postsegregational elimination of plasmid-less cells from the bacterial population (toxin–antitoxin system, TA). The additional genetic load includes (a) two highly related restriction-modification systems (R-M) (ORF9-ORF10 and ORF25-ORF26), (b) a putative novel catabolic operon and its regulator (SLF module), and (c) a putative retroelement (ORF8).
Replication system of pP62BP1
The replication systems (REP) of the vast majority of plasmids residing in gram-negative bacteria consist of (a) a gene encoding a replication initiation protein (Rep) and (b) a cis-required origin site (oriV), often containing iterons, i.e. directly repeated sequences representing Rep protein binding sites. Both of these elements could be distinguished within the REP region of pP62BP1, which has a structure typical for many theta-replicating plasmids.
The predicted RepB protein of pP62BP1 (ORF1; 305 amino acids; aa) shows the highest level of aa sequence identity (50%) with replication initiation proteins encoded by two Psychrobacter sp. strains, J466 and 1501(2011), isolated from the gut of the Atlantic herring (Clupea harengus) (Curson et al. 2010) and human blood, respectively (GenBank acc. nos. ACY02903 and EGK07200). A significant level of sequence similarity is also shared with a hypothetical protein (46%) of Kingella oralis ATCC 51147 (Betaproteobacteria; family Neisseriaceae) (GenBank acc. no. EEP66524) and replication proteins (30–35%) of several plasmids of Gammaproteobacteria: (a) p2ABSDF from Acinetobacter baumanii SDF (CAP02944), (b) pMMCU1 of Acinetobacter calcoaceticus (ACT83384), (c) pVSAL43 of Aliivibrio salmonicida (LFI1238; CAQ76621), (d) pALVIN01 of Allochromatium vinosum DSM 180 (ADC64139) and (e) pMBO-1 of Moraxella bovis Epp63 (BAD83730).
Inspection of the primary structure of the putative pP62BP1 RepB protein and its two closest homologs revealed the presence of two analogously located conserved regions, containing (a) a helix-turn-helix DNA binding motif (HTH) (residues 170–190 in RepB) (Dodd and Egan 1990) and (b) an N-terminal leucine zipper-like (LZ-like) motif that is probably responsible for dimerization of the Rep molecules (del Solar et al. 1998). The LZ-like motif of pP62BP1 RepB protein comprises 4 leucine residues separated by heptad intervals (Leu4, 11, 18, and 25).
Both the HTH and LZ-like motifs are also conserved in the well-characterized RepA protein of plasmid pPS10 from Pseudomonas syringae pv. savastonoi (Giraldo and Fernández-Tresguerres 2004), which shares around 32% identity with the putative polypeptide product of pP62BP1 ORF1. Analysis of the crystal structure of pPS10 RepA revealed the presence of two “winged-helix” domains (WH1 and WH2) (Giraldo et al. 2003), which play a crucial role in the control of initiation of plasmid replication (Gasset-Rosa et al. 2008). In silico analysis of the predicted secondary structure of the RepB of pP62BP1 (PredictProtein, YASPIN) suggests that this protein contains analogous WH domains in the regions covered by residues 45–120 and 125–230 (data not shown).
The putative oriV of pP62BP1 is located immediately upstream of repB (Fig. 2). This region contains six tandem repeat sequences of 20 bp (putative iterons, designated IT1–IT6), which begin 197 bases upstream of the predicted start codon (ATG) of repB. Iterons IT1–IT5 are identical, while IT6 differs from the others by 6 bases. It is noteworthy that the three 3′-terminal bases of IT6 are located within one of the inverted repeats that form a 12-bp-long palindromic sequence (Fig. 2b).

a Structure of the REP module of pP62BP1. A G+C content profile is shown in the upper panel with the relative positions of ORF33, oriV and repB indicated below (1 the arbitrary starting point of the plasmid’s nucleotide sequence). b Sequence organization of the proposed oriV. The dnaA box is shaded. Dashed arrows indicate A+T-rich sequences (DR1–DR4). Six iteron sequences (IT1–IT6) are marked by black arrows. The non-identical residues in the IT6 sequence are written in lowercase. Palindromic sequence is underlined. Additional repeats similar to the 3′-part of iterons (AR1–AR3) are indicated by gray arrows. The repB −35 and −10 promoter elements and a potential ribosome binding site (rbs) are boxed. The coding sequence of repB is marked by a large gray arrow in the background
The iteron-containing region of pP62BP1 (120 bp) is relatively G+C-rich (~42%) and is surrounded by two A+T-rich DNA segments (Fig. 2a). The one situated upstream of the iterons contains four adjacent A+T-rich direct repeats (DR1-DR4; consensus: 5′-ANT/CCT/CTGTTT-3′), which may function as a strand melting site during the initiation of plasmid replication (Fig. 2b). Upstream of DR1 there is a short sequence element (5′-TTATCCACA-3′) matching the consensus sequence recognized by a chromosomally encoded protein DnaA (5′-TTA/TTNCACA-3′; Messer 2002). The DnaA protein has been shown to participate in the replication initiation of several iteron-containing plasmids (del Solar et al. 1998). The A+T-rich region downstream of the iterons carries a predicted promoter sequence for repB. Its −35 box is surrounded by two imperfect inverted repeats, designated as AR (additional repeats; AR1: 5′-CTATAATGA-3′ and AR2: 5′-CTATGTAGA-3′), which share some similarity with the 3′-termini of the iterons (5′-CTAAATGACGC-3′). A third sequence of this type (AR3: 5′-TTTAAATGA-3′) was identified between the −10 TATA box and a putative ribosome binding site (rbs) of repB (Fig. 2b).
It is noteworthy that the organization of the putative oriV of pP62BP1 resembles that of pPS10, although the level of nucleotide sequence identity of these regions is rather low (data not shown). Plasmid pPS10 is a model iteron-containing, theta-replicating plasmid, whose replication has been thoroughly studied (Giraldo and Fernández-Tresguerres 2004). Based on the observed similarity between the Rep proteins and the shared structure of the oriV regions of pP62BP1 and pPS10, we assume that the replication control of these plasmids functions in a similar way, i.e. by binding of Rep proteins to the iterons or AR sequences, depending on the conformation of the proteins.
In contrast to pPS10, plasmid pP62BP1 contains an additional ORF (ORF33) which may constitute part of the REP region (Fig. 2a). ORF33 is located upstream of the putative oriV of pP62BP1 and predicted to encode a 69-aa protein containing a conserved HTH_XRE DNA binding domain (cd00093; residues 23–44) commonly found in many bacterial transcriptional regulators, including a number of TA system antitoxin proteins. The amino acid sequence of the predicted ORF33 protein shares the highest level of similarity with polypeptides encoded by several gammaproteobacteria, e.g. Vibrio cholerae 12129(1) and Enhydrobacter aerosaccus SK60 (64% identity; GenBank acc. nos. EEO01051 and EEV22138, respectively).
It is noteworthy that a protein of the XRE family (named RepA), which is highly related to ORF33, is also encoded within the REP region of the broad host range plasmid RA3 (IncU group). As in pP62BP1, the RA3 repA gene is located upstream of the ORF encoding the replication initiation protein RepB (which is not related to the Rep protein of pP62BP1). In the case of RA3, it has been demonstrated that expression of the repA gene is repressed by both the RepA and RepB proteins, which strongly suggests that the repA promoter can influence the regulation of repB expression (Kulinska et al. 2008). We hypothesize that a similar regulatory circuit may also occur in pP62BP1, but this has to be experimentally confirmed.
Stable maintenance systems
Partitioning system
Immediately upstream of the pP62BP1 REP module, two ORFs (ORF2 and ORF3) representing a putative partitioning system (PAR) were identified. Plasmid-encoded PAR systems, responsible for proper segregation of plasmid copies to daughter cells at cell division, are usually composed of two genes (often termed parA and parB) and a partitioning site where the ParB protein binds (Ebersbach and Gerdes 2005).
The predicted product of ORF2 contains a conserved ParA domain (cd02042) with a deviant Walker-A type ATPase motif adjacent to its N terminus (KGGSKGT, residues 10–16). The closest homologs of the ORF2 protein are encoded by genes in plasmid 1 of P. cryohalolentis K5 (97% identity) and in the chromosome of Enhydrobacter aerosaccus SK60 (80% identity).
ORF3 is placed downstream of ORF2 and encodes a putative protein with a ParG domain (PF09274). Moreover, the predicted secondary structure of the potential ORF3 product corresponds to the ribbon-helix-helix structure confirmed for the ParG domain-containing protein of plasmid pTP228 (Golovanov et al. 2003). Database searches revealed that the two most similar proteins (87 and 57% identity, respectively) are encoded by ORFs situated immediately downstream of the aforementioned ORF2 homologs from plasmid 1 of P. cryohalolentis K5 and E. aerosaccus. However, it is noteworthy that the coding sequence corresponding to ORF3 was not distinguished during annotation of the P. cryohalolentis K5 plasmid 1 sequence deposited in the GenBank database (acc. no. CP000324, position 21,128–21,331).
Taking into consideration (a) the predicted length of the polypeptides encoded within the par locus of pP62BP1, (b) the presence of a Walker-A type motif in the putative ParA (and absence of a HTH motif), and (c) the weak conservation of a predicted ParB aa sequence, it may be concluded that ORF2 and ORF3 represent the parA and parB genes of a type Ib partitioning system (Ebersbach and Gerdes 2005). The genes are separated by a short intergenic sequence (9 bp) and both are preceded by putative ribosome binding sites. Since the rbs of parB partially overlaps the stop codon of parA, it is highly probable that the two genes constitute an operon.
The third component of the putative type Ib par locus of pP62BP1, i.e. a centromere-like sequence parS, is thought to be located between the −10 TATA box and rbs of parA. This region is relatively A+T-rich and contains seventeen identical, adjacent 8-bp direct repeats (5′-AATACTCA-3′). Comparative analyses of nucleotide sequences upstream of genes encoding proteins with homology to ParA revealed that such sequence organization is apparently unique for pP62BP1. Interestingly, we found twelve 5′-AATACTCA-3′ repeats in the sequence of plasmid 1 of P. cryohalolentis K5. However, these repeats are not adjacent to the parA homolog, but form the 3′ terminus of a putative protein coding sequence Pcryo_2479 (see below).
It is also noteworthy that a stretch of eight 5′-TTTACTCA-3′ repeats similar to those present in the putative parS site of pP62BP1 was identified within the intergenic region between ORF27 and ORF28 (residues 31,259–31,298 bp). However, it is not known whether ParB binds to these sequences.
Toxin–antitoxin system
ORF23, one of the smallest among the identified coding sequences of pP62BP1, encodes a putative 62-aa polypeptide that belongs to the family of predicted HicA-like RNA binding proteins (COG1724). These proteins are thought to function as toxins of hicAB addiction systems, which are responsible for post-segregational elimination of plasmid-less cells from a bacterial population (Makarova et al. 2006; Jørgensen et al. 2009). The closest homologs of the ORF23 product (48–55% of identity) were identified in Caldicellulosiruptor obsidiansis OB47 (GenBank acc. no. ADL43535), C. saccharolyticus DSM 8903 (ABP65736) and Burkholderia xenovorans LB400 (ABE32795). Amino acid sequence alignments of these proteins gave a consensus which matches that for HicA-like proteins reported by Makarova et al. (2006). It is noteworthy that downstream of ORF23 we identified a short ORF22 encoding a putative 60-aa protein without any significant similarity to sequences deposited in open databases. However, corresponding short ORFs were also identified downstream of the aforementioned ORF23 homologs and their sequence alignment confirms some level of similarity to the HicB antitoxin consensus sequence (data not shown). Further analysis is required to determine whether ORF22 and ORF23 of pP62BP1 constitute a functional TA system.
Multimer resolution system
The amino acid sequence of the deduced ORF19 protein was found to contain a catalytic domain characteristic of the superfamily of serine recombinases (SR) which perform site-specific recombination of DNA molecules (cd00338). The enzymes of this group are functionally versatile and include integrases, resolvases, transposases and invertases (Smith and Thorpe 2002). The pP62BP1-encoded SR shares the highest level of similarity with a subgroup of these proteins harboring a SR_rep_par domain (cd03767). Typically, they function as resolvases in multimer resolution systems which represent a type of plasmid stabilization mechanism (Bahl et al. 2009).
Four amino acid sequences that exhibit more than 80% identity to the described SR were identified in Psychrobacter sp. 1501(2011) (GenBank acc. no. EGK07211), plasmid pRWF101 of Psychrobacter sp. PRwf-1 (ABQ95337), and in two strains of Acinetobacter baumanii: 6013150 (EGJ69339) and 6013113 (EGJ65601). Interestingly, careful analysis of the nucleotide sequence of plasmid 1 of P. cryohalolentis K5 identified two adjacent coding sequences whose products, designated Pcryo_2509 (ABE76286) and Pcryo_2479 (ABE76256), are similar to the N- and C-terminal parts of the pP62BP1-encoded SR, respectively. In silico joint translation of these two ORFs (including the intergenic sequence) produced an amino acid sequence 84% identical to that of the pP62BP1 ORF19 product (data not shown). We assume that a single transversion 5′-GAA-3′ → 5′-TAA-3′ resulting in a Glu → STOP ochre nonsense mutation might have been responsible for disrupting the original ORF19 homolog in the strain K5 plasmid. Moreover, in the 3′-terminal region of the Pcryo_2479 coding sequence, we discovered, to our surprise, 12 repeated sequences identical to those forming the putative parS of the pP62BP1 partitioning system (see above).
It is noteworthy that ORF19 is flanked by 83-bp-long inverted repeats, IRLRES and IRRRES, that are 77% identical (different in 20 nt positions). IRLRES ends 22 nt upstream of the gene’s putative START codon, while IRRRES contains its termination codon. Comparative analyses confirmed that similar sequences occur in several replicons of Psychrobacter spp., including the chromosomes of P. cryohalolentis K5, P. arcticus 273-4 and two Psychrobacter sp. strains, 1501(2011) and PRwf-1; while in the aforementioned plasmid 1 of P. cryohalolentis and pRWF101, single copies of the IR are found upstream of the ORFs encoding the pP62BP1 SR homologs.
Additional genetic load
Restriction-modification systems
Sequences encoding two restriction-modification systems, designated R-M1 (ORF9-ORF10) and R-M2 (ORF25-ORF26), were identified in pP62BP1. Both consist of genes encoding a methyltransferase (MTase) and a restriction endonuclease (REase) placed tail to tail with overlapping termination codons. Interestingly, these two systems (separated by ca. 17 kbp) show a very high level of identity, since the nucleotide sequences of their corresponding components differ only in 33 nt (MTase genes) and 16 nt (REase genes). Consequently, the level of amino acid identity of the potential MTases is 99% (420/424 aa), and that of the putative REases is 96% (250/261 aa).
Sequence comparisons revealed that the putative MTases encoded by ORF9 and ORF25 are highly similar to numerous C5-cytosine-specific DNA methylases, encoded by, e.g. Vibrio sp. RC586 (69% identity; GenBank acc. no. EEY99607), Acinetobacter baumanii ATCC 19606 (66%; EEX04797), and Lactococcus lactis subsp. cremoris UC503 (55%; AAB66696). The recognition sequence of these enzymes is predicted to be 5′-CCNGG-3′ (methylated cytosine underlined).
The predicted amino acid sequences of the protein products of ORF9 and ORF25 contain six motifs conserved among C5-methyltransferases (Pósfai et al. 1989) (data not shown). Sequence conservation of the potential target recognition domains (TRDs) of the pP62BP1-encoded MTases and their homologs indicates that they are likely to recognize the same 5′-CCNGG-3′ sequence. Furthermore, a MUSCLE alignment of 93 characterized and putative cytosine C5-methyltransferases with the same recognition sequence deposited in REBASE (data not shown) suggested that conservation of the amino acid string G(K/N)GFx13T(I/L)x3YYK in their TRDs may be characteristic of 5′-CCNGG-3′ MTases (cf. Neely and Roberts 2008). This motif corresponds to residues 316–340 in the ORF9 and ORF25 products. Moreover, within the N-terminal regions of these proteins, we identified a HTH_XRE domain (cd00093), which may be responsible for DNA–protein interactions other than the recognition of a methylation site, as was experimentally proved for the aforementioned enzyme M.ScrFI of Lactococcus lactis subsp. cremoris UC503 (Butler and Fitzgerald 2001).
The potential proteins encoded by ORF10 and ORF26 belong to the LlamI family of type II REases and so are predicted to cleave DNA after the first cytosine in the sequence 5′-CCNGG-3′. Only five proteins with significant homology to the pP62BP1 REases were found in the open databases. All of these are encoded in the vicinity of the aforementioned MTase homolog genes in Lachnospiraceae bacterium 5_1_63FAA (51% identity; GenBank acc. no. EFV17523), V. parahaemolyticus AQ4037 (48%; EFO45071), P. marinus NATL1A (46%; ABM74602), and L. lactis subsp. cremoris strains U503 (46%; AAB66695) and M19 (45%; AAM03111) [NB. the methyltransferase sequences of these two strains are identical (Szatmari et al. 2006)]. In the amino acid sequences of all these putative REases we identified a bipartite catalytic motif PD–(D/E)XK (residues 56–64 in the pP62BP1 REases) (Orlowski and Bujnicki 2008). Notably, the 4-aa-long spacer sequence between the conserved PD and (D/E)XK amino acid strings is unusually short (Kosinski et al. 2007).
The amino acid sequences of two of the aforementioned REases from L. lactis were used by Kosinski et al. (2007) to construct a protein–DNA interaction model of the R.MvaI enzyme, which was partially confirmed following determination of the crystal structure of this complex (PDB: 2OA9_A) (Kaus-Drobek et al. 2007). We performed a comparative analysis of the amino acid sequences of R.MvaI and pP62BP1 REases and, in spite of the low level of sequence identity (13% identity, 34/241 aa), analogies in the secondary structures of these proteins were observed. Moreover, seven amino acids of proven importance to the functionality of R.MvaI are conserved in the ORF10 and ORF26 products. These residues presumably take part in (a) the interaction with the phosphate backbone of DNA (residues K101, K183), (b) the recognition of C and G residues within the restriction site (D226, R241), and (c) the complexing of Mg2+ in the catalytic site of the enzyme (D57, E62, K64).
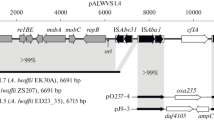
The aforementioned tail to tail organization of the ORFs of the pP62BP1 R-M1 and R-M2 systems with overlapping of their 3′ ends is rather uncommon among the restriction-modification systems (Wilson and Murray 1991). A comparison with the four most closely related systems described above revealed that tail to tail orientation of the component genes exists only in the case of P. marinus NATL1A (Fig. 3). Each of the three other homologous systems contains an additional methyltransferase gene as well as an ORF encoding a putative protein of unknown function (COG4933), neither of which is present in pP62BP1 or P. marinus.

Comparison of the genetic structures of R-M systems encoding proteins homologous to the enzymes of the R-M systems harbored by pP62BP1. Organism names, the ranges of the compared sequences and their GenBank acc. nos. are shown on the right. (NB Only one genetic structure is shown for pP62BP1, representing both R-M1 and R-M2.) ORFs are marked by block arrows colored according to the proposed function of their product (see legend). The GenBank acc. nos. of the encoded proteins are given above or below each ORF. The percentage values indicate the level of identity between proteins encoded by corresponding ORFs, which are paired by shaded regions
The putative slf operon
The region of the pP62BP1 sequence located between R-M1 and R-M2 was found to contain a putative phenotypic module comprising nearly 25% of the plasmid genome. This is composed of five ORFs, four of which (slfL, slfS, slfH, slfC) are tandemly oriented, with the fifth (slfR) located upstream of the others in the reverse transcriptional orientation (Fig. 4a). Analysis of the possible functions of the encoded proteins suggested that they may enable the bacterial host to metabolize organic sulfates present in the environment.

a Genetic organization of the pP62BP1 SLF module. ORFs are represented by block arrows. The location and orientation of predicted promoter sequences (P) for slfC and slfR are indicated by arrows. b Proposed metabolic pathway comprising reactions catalyzed by the enzymes encoded in the slf operon, with dodecyl sulfate as the starting substrate (see text for details). Dashed arrow indicates transport into the cell. The proposed enzyme names are based on their functions: SlfS, alkyl sulfatase; SlfH and SlfC, dehydrogenases catalyzing oxidation of hydroxyl and carbonyl groups, respectively; SlfL, fatty-acid-CoA ligase
The crucial enzyme in this process—an alkylsulfatase—is a putative product of the ORF designated slfS. Sulfatases (EC 3.1.6-) constitute a large and heterogeneous group of enzymes which catalyze the hydrolysis of organic esters of sulfuric acid to produce the corresponding alcohol and hydrogen sulfate (Hanson et al. 2004). Three types of sulfatases have been distinguished to date: (1) predominantly eukaryotic arylsulfatases, (2) sulfatases belonging to the Fe2+ α-ketoglutarate-dependent dioxygenase superfamily, and (3) sulfatases related to metallo-β-lactamases (Long et al. 2011). Comparative analysis allowed us to classify SlfS as a type 3 sulfatase. The putative protein shares extensive amino acid sequence homology with numerous enzymes of Pseudomonas spp; in particular, a considerable level of identity (57%) was observed with the well-characterized SdsA1 of P. aeruginosa PAO1 (Hagelueken et al. 2006). Amino acid sequence alignment of SlfS and SdsA1 revealed their similar domain organization (data not shown). A 21-aa-long signal peptide was identified at the N terminus of SlfS (PrediSi), which corresponds to the 19-aa sequence of SdsA1 that was shown to direct secretion of this enzyme. Both proteins harbor (a) a Zn2+-binding motif (THxHxDHxGGx102 Ex18AEx44 H), which forms part of a metallo-β-lactamase-like domain, (b) a C-terminal SCP-2 domain, and (c) a central dimerization domain.
The four other ORFs of the pP62BP1 SLF module encode putative proteins whose closest homologs were found in Acinetobacter johnsonii SH046 (see Table 1). Detailed bioinformatic analyses identified the slfL product as a fatty-acid-CoA synthetase (FACS; EC 6.2.1.3), which belongs to the adenylate-forming enzyme superfamily (Black and DiRusso 2003). Two characteristic motifs were found in this protein: an ATP-AMP motif YTSGTTGxPKGVx125-130GYGxTE, conserved in all the enzymes of the superfamily and a FACS signature motif DGWLHTGDIGxWxPxGxLKIIDRKK (Black et al. 1997). In silico analyses indicated that SlfL is closely related to the FACS enzymes specific for long- or medium-chain fatty acids (NCBI Conserved Domains Search). The ligation of these substrates with CoA, presumably catalyzed by SlfL, results in the production of acyl-CoA, which may subsequently become a substrate for β-oxidation or the biosynthesis of other lipids.
The segment of the SLF module that contains slfH, slfC and slfR was found to be equivalent to a region of the A. johnsonii SH046 chromosome (see Table 1). The conserved domains GMC_oxred_N and GMC_oxred_C, which are characteristic of GMC oxidoreductases, were identified in the deduced amino acid sequence of SlfH (Cavener 1992). Since the GMC_oxred_N domain binds FAD as a cofactor, these enzymes appear to belong to the flavoprotein superfamily (Fraaije and Mattevi 2000). The two domains are also found in long-chain alcohol dehydrogenases (EC 1.1.3.20), so SlfH might catalyze their oxidation to the corresponding aldehyde. The putative SlfC protein is predicted to exhibit similar oxidoreductase activity, although with specificity for aldehydes (Pfam: PF00171). This enzyme is likely to require NAD+ or NADP+ as a cofactor.
We propose a chain of reactions catalyzed by the four identified enzymes leading from alkylsulfate to acyl-CoA (Fig. 4). Secreted SlfS hydrolyses the alkyl sulfate and the produced alcohol (after being transported into the bacterial cell) is oxidized to a fatty acid, which, in turn, is activated by SlfL-mediated ligation with CoA. The presence of a similar metabolic pathway for the biodegradation of dodecyl sulfate (SDS) by Pseudomonas sp. C12B was confirmed by Thomas and White (1989). Using the 14C-radiotracer technique they observed that ~70% of the radiolabel was released as 14CO2 as a result of β-oxidation, while the remainder was incorporated into cellular components such as membrane lipids.
The exact nature of the starting substrate for the proposed pathway remains an open question. In a preliminary experiment we confirmed that Psychrobacter sp. DAB_AL62B can use SDS as the sole source of carbon and energy (data not shown). However, it seems unlikely that this anthropogenic compound represents the main substrate for alkylsulfatase in the Arctic environment, unless it is a contaminant from scientific research, the fishing industry or tourism (George 2002). Hagelueken et al. (2006) suggested that the specificity of SdsA1 (a homolog of the pP62BP1-encoded SlfS) towards SDS is incidental and that sulfated sugars such as sulfoglycolipids may represent its physiological substrates. Such compounds have been detected in the salt glands of sea birds (Ishizuka 1997), so it is not unlikely that they are present in the environment from which the pP62BP1 host strain was isolated. Furthermore, the ability to directly use alkyl chains in the synthesis of membrane elements, confirmed by Thomas and White (1989), may be of adaptive importance to psychrophilic bacteria. It is known that the modulation of fatty acid composition is a common mechanism for maintaining the correct membrane fluidity at low temperatures (Wang et al. 2009).
The genetic organization of the region containing the ORFs encoding the aforementioned enzymes suggests that they are arranged in an operon. It is noteworthy that although the reactions comprising the proposed metabolic pathway have been long known, we are unaware of any previous reports of a similarly structured gene cluster. In addition, we predict that the activity of the operon is controlled by a transcriptional regulator SlfR. This protein belongs to the AraC/XylS family that includes a large number of activators involved in carbon metabolism, stress response and virulence (Egan 2002). The conserved domains Arabinose_bd (PF00165) and HTH_AraC (PF12625) were identified in the N- and C-terminal regions of SlfR, respectively. The latter contains two helix-turn-helix motifs characteristic of AraC/XylS family proteins, while the N-terminal domain is most probably responsible for effector binding and dimerization of the regulator (Domínguez-Cuevas et al. 2008).
Typically, the mechanism of transcription control by AraC/XylS family regulators involves binding to specific DNA sequences within cognate promoters, which results in positive stimulation of gene expression. This depends on the recognition of a specific effector that causes a conformational change in the regulator and consequently its correct positioning at the binding sites (Gallegos et al. 1997). We assume that the identified SlfR protein may interact with series of sequence repeats which overlap the putative promoters of slfC and slfR. Interestingly, among these we identified a pair of direct repeats, 5′-TTGA(N6)AGGACA-3′, which have a similar structure to the XylS binding sites (Domínguez-Cuevas et al. 2010). Corresponding repeated sequences are also present upstream of ORFs encoding the closest homologs of SlfR from A. johnsonii SH046 (GenBank acc. no. EEY97636), Aliivibrio salmonicida LFI1238 (CAQ81021) and Shewanella sp. ANA-3 (ABK46545).
The organization of the intergenic sequence between slfC and slfR suggests that the proposed binding of the regulator may affect transcription from both promoters. According to our prediction, the mechanisms of SlfR activity may resemble the model proposed for Pseudomonas putida XylS protein (Domínguez-Cuevas et al. 2008). If this is the case, SlfR should bind to specific sequences in the presence of an effector and thus promote the transcription from the slfC promoter, possibly simultaneously autoregulating its own expression. Functional analysis will be necessary to elucidate the exact nature of this process, including identification of the effector molecule and examination of the possible existence of secondary promoters or other regulatory sequences.
Other ORFs
More than 50% of the ORFs of pP62BP1 do not belong to the aforementioned genetic modules (see Table 1; Fig. 1). Observed sequence similarities allowed us to predict the functions of putative proteins encoded by only three of the ORFs (ORF8, ORF11, and ORF26).
Detailed analysis of the amino acid sequence of the deduced ORF8 polypeptide suggested that it is related to reverse transcriptases (RT) of the RVT_1 family (PF00078), most often encoded by retroelements such as retrons or group II introns (Lampson et al. 2005). Within the putative protein sequence we identified seven conserved domains characteristic of RT (Xiong and Eickbush 1990), including domain 5 that contains a YADD motif comprising the enzyme active site (residues 281–284; Zimmerly et al. 2001). The presence of this ORF suggests that pP62BP1 carries a retroelement, most probably a retron.
In the N-terminal part of the putative 651-aa-long product of ORF11 we found a relatively short region (residues 23–69) with homology to the consensus sequence of the conserved domain of MutL-like proteins (COG0323). Members of this large protein family are involved in DNA mismatch repair and have been found in all domains of life (Yang 2000). However, the aforementioned limited homology is insufficient to assume a similar function for the ORF11 product.
In the sequence of the putative ORF27 polypeptide we found several motifs which suggest ATPase activity, including a Walker A motif in its N-terminal region, a Walker B motif, a D-loop and an H-loop in its central region. These features occur in a large group of proteins which harbor ATP-binding cassettes (ABC; cd00267). This superfamily comprises, among others, ABC transporters, DNA repair proteins (e.g. UvrA, MutS and Rad50) and proteins involved in the structural maintenance of chromosomes (SMC) (Hopfner and Tainer 2003). Bioinformatic analyses of the amino acid sequence encoded by ORF27 distinguished extensive overlapping segments that exhibit significant similarity to the conserved domains of OLD-type endonucleases and SMC proteins. The former enzymes are engaged in DNA replication, recombination and repair (COG3593), while the latter proteins are crucial for chromosome dynamics including their condensation and segregation during cell division (Graumann and Knust 2009). In the absence of supporting experimental evidence, it is not possible to predict the function of the putative ORF27 protein.
Comparative analysis of pP62BP1 and other plasmids of Psychrobacter spp.
A comparative analysis of the nucleotide sequences of pP62BP1 and other plasmids of Psychrobacter spp. revealed significant levels of identity between certain sequence segments only in the case of the two largest plasmids known to date: plasmid 1 of P. cryohalolentis K5 (Fig. 5) and pRWF-101 of Psychrobacter sp. PRwf-1.

Linear alignment of pP62BP1 and plasmid 1 of Psychrobacter cryohalolentis K5. ORFs are marked by block arrows. In the strain K5 plasmid, several GenBank acc. nos. are given for reference, while the ORF whose sequence has not been deposited in GenBank is encircled. In pP62BP1, the defined genetic modules are boxed (see Fig. 1 for the abbreviations used). Vertical black bars mark the positions of 142-nt-long inverted repeats (IRL and IRR; see text for details). Regions of the plasmids sharing >70% identity (E value >1e−10) are connected by black connecting lines; those corresponding to pP62BP1 iterons and the parS sequence are marked accordingly. The scale is indicated by the bar in the lower right-hand corner
Highly similar ORFs presumed to encode serine recombinases (putative MRS systems) are present in all three replicons. However, as stated above, the sequence from the strain K5 plasmid appears to be disrupted and considerably rearranged, since the region corresponding to pP62BP1 parS is situated at its 3′ terminus. The presence of homologous PAR loci and iteron repeats in both of these replicons indicates some relationship between them, although it appears to have been obscured by genetic rearrangements that occurred in the evolution of the P. cryohalolentis K5 plasmid.
Other corresponding sequences are found within two regions of pP62BP1 which encompass a pair of 142-bp-long inverted repeats (80% identity; positions 11,496–11,637 and 31,055–31,196) (Fig. 5). Since similar sequences are also present in the chromosomes of Psychrobacter sp. PRwf-1, P. cryohalolentis K5 and P. arcticus 273-4, we assume that they might be associated with site-specific recombination or lateral gene transfer events that could have influenced the evolution of Psychrobacter spp. replicons.
Conclusions
To date, pP62BP1 is the largest plasmid of Psychrobacter spp. for which a comprehensive analysis of its nucleotide sequence has been reported. In this study, we have used in silico methods to describe in detail the structure and putative functions of the genetic modules carried by this plasmid. The possible regulatory mechanisms of genes comprising several of these modules have also been proposed. We found that the compact backbone region of pP62BP1 consists of genes and regulatory sequences associated with its replication and stable inheritance (REP and PAR). Preliminary studies have confirmed that these modules are functional in other Psychrobacter spp. strains (data not shown), and we predict that they could be used to develop shuttle vectors for bacteria of this genus.
The two nearly identical restriction-modification systems harbored by pP62BP1 seem to be a feature that is unique among bacterial plasmids analyzed so far. There are two plausible non-mutually exclusive reasons why both R-M1 and R-M2 are stably maintained in this plasmid. First, they may function as a type of postsegregational cell killing system that increases stability of the plasmid in the bacterial population (Ichige and Kobayashi 2005). Second, a recombination event leading to the loss of one system or the production of a hybrid system could destabilize the putative common regulatory circuit controlling the expression of the R-M1- and R-M2-encoded genes. Further functional analyses of this plasmid may help to increase our understanding of this R-M regulatory mechanism and the role of these systems in prokaryotic genomes.
Likewise, a putative catabolic operon responsible for the transformation of organic sulfate esters, similar to that carried by pP62BP1, has not been described before. It is likely that possession of this operon allows the host strain to adapt to environmental conditions, presumably by conferring the ability to utilize a specific class of compounds for energy production and/or biosyntheses. The regulatory elements that stimulate the transcriptional activity of the slf operon have the potential to be used in the construction of vectors for a novel inducible protein expression system.
In summary, the findings of this study greatly expand our knowledge of plasmids of the psychrophilic bacteria as well as their possible role in the lateral gene transfer and the adaptation of microorganisms to specific Arctic environments. The presented analyses identify a number of interesting avenues to be explored in further experimental studies.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Bahl MI, Hansen LH, Sørensen SJ (2009) Persistence mechanisms of conjugative plasmids. Methods Mol Biol 532:73–102
Black PN, DiRusso CC (2003) Transmembrane movement of exogenous long-chain fatty acids: proteins, enzymes, and vectorial esterification. Microbiol Mol Biol Rev 67:454–472
Black PN, Zhang Q, Weimar JD, DiRusso CC (1997) Mutational analysis of a fatty acyl-coenzyme A synthetase signature motif identifies seven amino acid residues that modulate fatty acid substrate specificity. J Biol Chem 272:4896–4903
Butler D, Fitzgerald GF (2001) Transcriptional analysis and regulation of expression of the ScrFI restriction-modification system of Lactococcus lactis subsp. cremoris UC503. J Bacteriol 183:4668–4673
Cavener DR (1992) GMC oxidoreductases. A newly defined family of homologous proteins with diverse catalytic activities. J Mol Biol 223:811–814
Cavicchioli R, Siddiqui KS, Andrews D, Sowers KR (2002) Low-temperature extremophiles and their applications. Curr Opin Biotechnol 13:253–261
Cieśliński H, Werbowy K, Kur J, Turkiewicz M (2008) Molecular characterization of a cryptic plasmid from the psychrotrophic antarctic bacterium Pseudoalteromonas sp. 643A. Plasmid 60:154–158
Claudel-Renard C, Chevalet C, Faraut T, Kahn D (2003) Enzyme-specific profiles for genome annotation: PRIAM. Nucleic Acids Res 31:6633–6639
Curson AR, Sullivan MJ, Todd JD, Johnston AW (2010) Identification of genes for dimethyl sulfide production in bacteria in the gut of Atlantic Herring (Clupea harengus). ISME J 4:144–146
del Solar G, Giraldo R, Ruiz-Echevarría MJ, Espinosa M, Díaz-Orejas R (1998) Replication and control of circular bacterial plasmids. Microbiol Mol Biol Rev 62:434–464
Dodd IB, Egan JB (1990) Improved detection of helix-turn-helix DNA-binding motifs in protein sequences. Nucleic Acids Res 18:5019–5026
Domínguez-Cuevas P, Marín P, Marqués S, Ramos JL (2008) XylS-Pm promoter interactions through two helix-turn-helix motifs: identifying XylS residues important for DNA binding and activation. J Mol Biol 375:59–69
Domínguez-Cuevas P, Ramos JL, Marqués S (2010) Sequential XylS-CTD binding to the Pm promoter induces DNA bending prior to activation. J Bacteriol 192:2682–2690
Duilio A, Tutino ML, Matafora V, Sannia G, Marino G (2001) Molecular characterization of a recombinant replication protein (Rep) from the Antarctic bacterium Psychrobacter sp. TA144. FEMS Microbiol Lett 198:49–55
Ebersbach G, Gerdes K (2005) Plasmid segregation mechanisms. Annu Rev Genet 39:453–479
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Egan SM (2002) Growing repertoire of AraC/XylS activators. J Bacteriol 184:5529–5532
Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A (2010) The Pfam protein families database. Nucleic Acids Res 38:D211–D222
Fraaije MW, Mattevi A (2000) Flavoenzymes: diverse catalysts with recurrent features. Trends Biochem Sci 25:126–132
Gallegos MT, Schleif R, Bairoch A, Hofmann K, Ramos JL (1997) Arac/XylS family of transcriptional regulators. Microbiol Mol Biol Rev 61:393–410
Gao Y, Mathee K, Narasimhan G, Wang X (1999) Motif detection in protein sequences. In: Proceedings of the 6th SPIRE conference, pp 63–72
Gasset-Rosa F, Maté MJ, Dávila-Fajardo C, Bravo J, Giraldo R (2008) Binding of sulphonated indigo derivatives to RepA-WH1 inhibits DNA-induced protein amyloidogenesis. Nucleic Acids Res 36:2249–2256
George AL (2002) Seasonal factors affecting surfactant biodegradation in Antarctic coastal waters: comparison of a polluted and pristine site. Mar Environ Res 53:403–415
Giraldo R, Fernández-Tresguerres ME (2004) Twenty years of the pPS10 replicon: insights on the molecular mechanism for the activation of DNA replication in iteron-containing bacterial plasmids. Plasmid 52:69–83
Giraldo R, Fernández-Tornero C, Evans PR, Díaz-Orejas R, Romero A (2003) A conformational switch between transcriptional repression and replication initiation in the RepA dimerization domain. Nat Struct Biol 10:565–571
Golovanov AP, Barillà D, Golovanova M, Hayes F, Lian LY (2003) ParG, a protein required for active partition of bacterial plasmids, has a dimeric ribbon-helix-helix structure. Mol Microbiol 50:1141–1153
Graumann PL, Knust T (2009) Dynamics of the bacterial SMC complex and SMC-like proteins involved in DNA repair. Chromosome Res 17:265–275
Hagelueken G, Adams TM, Wiehlmann L, Widow U, Kolmar H, Tümmler B, Heinz DW, Schubert WD (2006) The crystal structure of SdsA1, an alkylsulfatase from Pseudomonas aeruginosa, defines a third class of sulfatases. Proc Natl Acad Sci USA 103:7631–7636
Hanson SR, Best MD, Wong CH (2004) Sulfatases: structure, mechanism, biological activity, inhibition, and synthetic utility. Angew Chem Int Ed Engl 43:5736–5763
Hiller K, Grote A, Scheer M, Münch R, Jahn D (2004) PrediSi: prediction of signal peptides and their cleavage positions. Nucleic Acids Res 32:W375–W379
Hopfner KP, Tainer JA (2003) Rad50/SMC proteins and ABC transporters: unifying concepts from high-resolution structures. Curr Opin Struct Biol 13:249–255
Ichige A, Kobayashi I (2005) Stability of EcoRI restriction-modification enzymes in vivo differentiates the EcoRI restriction-modification system from other postsegregational cell killing systems. J Bacteriol 187:6612–6621
Ishizuka I (1997) Chemistry and functional distribution of sulfoglycolipids. Prog Lipid Res 36:245–319
Jørgensen MG, Pandey DP, Jaskolska M, Gerdes K (2009) HicA of Escherichia coli defines a novel family of translation-independent mRNA interferases in bacteria and archaea. J Bacteriol 191:1191–1199
Kaus-Drobek M, Czapinska H, Sokołowska M, Tamulaitis G, Szczepanowski RH, Urbanke C, Siksnys V, Bochtler M (2007) Restriction endonuclease MvaI is a monomer that recognizes its target sequence asymmetrically. Nucleic Acids Res 35:2035–2046
Kosinski J, Kubareva E, Bujnicki JM (2007) A model of restriction endonuclease MvaI in complex with DNA: a template for interpretation of experimental data and a guide for specificity engineering. Proteins 68:324–336
Kulinska A, Czeredys M, Hayes F, Jagura-Burdzy G (2008) Genomic and functional characterization of the modular broad-host-range RA3 plasmid, the archetype of the IncU group. Appl Environ Microbiol 74:4119–4132
Lampson BC, Inouye M, Inouye S (2005) Retrons, msDNA, and the bacterial genome. Cytogenet Genome Res 110:491–499
Lin K, Simossis VA, Taylor WR, Heringa J (2005) A simple and fast secondary structure prediction algorithm using Hidden Neural Networks. Bioinformatics 21:152–159
Long M, Ruan L, Li F, Yu Z, Xu X (2011) Heterologous expression and characterization of a recombinant thermostable alkylsulfatase (sdsAP). Extremophiles 15:293–301
Makarova KS, Grishin NV, Koonin EV (2006) The HicAB cassette, a putative novel, RNA-targeting toxin–antitoxin system in archaea and bacteria. Bioinformatics 22:2581–2584
Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, Deweese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH (2011) CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res 39:D225–D229
Messer W (2002) The bacterial replication initiator, DnaA; DnaA and oriC, the bacterial mode to initiate DNA replication. FEMS Microbiol Rev 26:355–374
Miyake R, Kawamoto J, Wei YL, Kitagawa M, Kato I, Kurihara T, Esaki N (2007) Construction of a low-temperature protein expression system using a cold-adapted bacterium, Shewanella sp. strain Ac10, as the host. Appl Environ Microbiol 73:4849–4856
Narasimhan G, Bu C, Gao Y, Wang X, Xu N, Mathee K (2002) Mining for motifs in protein sequences. J Comput Biol 9:707–720
Neely RK, Roberts RJ (2008) The BsaHI restriction-modification system: cloning, sequencing and analysis of conserved motifs. BMC Mol Biol 9:48–57
Orlowski J, Bujnicki JM (2008) Structural and evolutionary classification of Type II restriction enzymes based on theoretical and experimental analyses. Nucleic Acids Res 36:3552–3569
Pagni M, Ioannidis V, Cerutti L, Zahn-Zabal M, Jongeneel CV, Hau J, Martin O, Kuznetsov D, Falquet L (2007) MyHits: improvements to an interactive resource for analyzing protein sequences. Nucleic Acids Res 35:W433–W437
Parrilli E, De Vizio D, Cirulli C, Tutino ML (2008) Development of an improved Pseudoalteromonas haloplanktis TAC125 strain for recombinant protein secretion at low temperature. Microb Cell Fact 7:2
Pósfai J, Bhagwat AS, Pósfai G, Roberts RJ (1989) Predictive motifs derived from cytosine methyltransferases. Nucleic Acids Res 17:2421–2435
Roberts RJ, Vincze T, Posfai J, Macelis D (2010) REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res 38:D234–D236
Rodrigues DF, da C Jesus E, Ayala-Del-Río HL, Pellizari VH, Gilichinsky D, Sepulveda-Torres L, Tiedje JM (2009) Biogeography of two cold-adapted genera: Psychrobacter and Exiguobacterium. ISME J 3:658–665
Rost B, Yachdav G, Liu J (2003) The PredictProtein Server. Nucleic Acids Res 32:W321–W326
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B (2000) Artemis: sequence visualization and annotation. Bioinformatics 16:944–945
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Smith MC, Thorpe HM (2002) Diversity in the serine recombinases. Mol Microbiol 44:299–307
Szatmari G, Hua NM, Vzdornov D, Daigle F, Smoragiewicz W, Mamet-Bratley MD, Karska-Wysocki B (2006) In vitro expression of the restriction endonucleases LlaMI and ScrFI isolated from Lactococcus lactis M19 and UC503. J Biotechnol 121:144–153
Thomas OR, White GF (1989) Metabolic pathway for the biodegradation of sodium dodecyl sulfate by Pseudomonas sp. C12B. Biotechnol Appl Biochem 11:318–327
Tutino ML, Duilio A, Moretti MA, Sannia G, Marino G (2000) A rolling-circle plasmid from Psychrobacter sp. TA144: evidence for a novel rep subfamily. Biochem Biophys Res Commun 274:488–495
Wang F, Xiao X, Ou HY, Gai Y, Wang F (2009) Role and regulation of fatty acid biosynthesis in the response of Shewanella piezotolerans WP3 to different temperatures and pressures. J Bacteriol 191:2574–2584
Werbowy K, Cieśliński H, Kur J (2009) Characterization of a cryptic plasmid pSFKW33 from Shewanella sp. 33B. Plasmid 62:44–49
Wilson GG, Murray NE (1991) Restriction and modification systems. Annu Rev Genet 25:585–627
Xiong Y, Eickbush TH (1990) Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J 9:3353–3362
Yang W (2000) Structure and function of mismatch repair proteins. Mutat Res 460:245–256
Zhao DL, Yu ZC, Li PY, Wu ZY, Chen XL, Shi M, Yu Y, Chen B, Zhou BC, Zhang YZ (2011) Characterization of a cryptic plasmid pSM429 and its application for heterologous expression in psychrophilic Pseudoalteromonas. Microb Cell Fact 10:30
Zimmerly S, Hausner G, X-c Wu (2001) Phylogenetic relationships among group II intron ORFs. Nucleic Acids Res 29:1238–1250
Acknowledgments
We acknowledge M. Zmuda-Baranowska, M. Zdanowski and P. Borsuk for providing Psychrobacter sp. DAB_AL62B. This work was supported by the Ministry of Science and Higher Education, Poland (Grant No. NN303 816340).
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by L. Huang.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lasek, R., Dziewit, L. & Bartosik, D. Plasmid pP62BP1 isolated from an Arctic Psychrobacter sp. strain carries two highly homologous type II restriction-modification systems and a putative organic sulfate metabolism operon. Extremophiles 16, 363–376 (2012). https://doi.org/10.1007/s00792-012-0435-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-012-0435-2