
Abstract
Formaldehyde ferredoxin oxidoreductase from Pyrococcus furiosus is a homotetrameric protein with one tungstodipterin and one [4Fe–4S] cubane per 69-kDa subunit. The enzyme kinetics have been studied under steady-state conditions at 80 °C and pre-steady state conditions at 50 °C, in the latter case via monitoring of the relatively weak (ε ≈ 2 mM−1 cm−1) optical spectrum of the tungsten cofactor. The steady-state data are consistent with a substrate substituted-enzyme mechanism for three substrates (formaldehyde plus two ferredoxin molecules). The K M value for free formaldehyde (21 μM) with ferredoxin as an electron acceptor is approximately 3 times lower than the value measured when benzyl viologen is used as an acceptor. The K M of ferredoxin (14 μM) is an order of magnitude less than previously reported values. An explanation for this discrepancy may be the fact that high concentrations of substrate are inhibitory and denaturing to the enzyme. Pre-steady-state difference spectra reveal peak shifts and a lack of isosbestic points, an indication that several processes happen in the first seconds of the reaction. Two fast processes (k obs1 = 4.7 s−1, k obs2 = 1.9 s−1) are interpreted as oxidation of the substrate followed by rearrangement of the active site. Alternatively, these processes could be the entry/binding of the substrate followed by its oxidation. The release of the product and the electron shuffling over the tungsten and iron–sulfur center in the absence of an external electron acceptor are slower (k obs3 = 6.10 × 10−2 s−1, k obs4 = 2.18 × 10−2 s−1). On the basis of these results in combination with results from previous electron paramagnetic resonance studies an activation route plus catalytic redox cycle is proposed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tungstoenzymes and molybdoenzymes typically catalyze the generic n = 2 reaction: R + H2O ↔ RO + 2[H], where R is either an oxoanion (e.g., sulfite) or a relatively small organic metabolite (e.g., an aldehyde). On the basis of structural (cofactor and protein) and functional properties molybdoenzymes are classified in three families: the sulfite oxidase family, the xanthine oxidase family, and the dimethyl sulfoxide reductase (DMSOR) family [1]. The latter also encompasses a limited number of tungsten-containing formate dehydrogenases. The majority of the tungstoenzymes, however, cannot be classified in any of these three families; they form a separate family of aldehyde oxidoreductases (AORs) together with a few apparently less common AOR molybdoenzyme members [2]. The mechanism of action of molybdoenzymes has been extensively studied for several decades, especially for xanthine oxidase [3]. In contrast, attempts to understand the reaction mechanism of tungstoenzymes, in particular for the members of the AOR family, are more recent and of limited extent, although the available structural information on tungstoenzymes and that on molybdoenzymes are comparable. The present study leads to the first proposal of a complete catalytic cycle of the AOR family member formaldehyde oxidoreductase (FOR).
The cofactor common to the enzymes of all four families consists of a single Mo(VI/IV) or W(VI/IV) ion coordinated by the dithioleno sulfurs of one or two three-ring pterin ligands sometimes with an additional nucleotide attached through a phosphoester bond [4]. All tungsten systems appear to be nonnucleotide dipterin systems with an additional magnesium phosphate coordination between the pterins [5, 6]. The coordination number of the Mo or W is 4–7; extra ligands are provided by an amino acid side chain and/or by O,S of a small, nonprotein ligand. The vast majority of the Mo/W enzymes have additional prosthetic groups for interaction with a second substrate (e.g., NADH) and/or for electron transfer with a protein natural redox partner (e.g., ferredoxin). These heme, flavin, and/or iron–sulfur prosthetic groups are strongly colored to the extent that optical monitoring of the relatively weakly colored (see below) metallopterin has thus far not been tried for mechanistic studies of these complex enzymes. Electron paramagnetic resonance (EPR) spectroscopy is not a competitive alternative, because the only EPR-detectable oxidation state, Mo(V) or W(V), does not appear to be a competent intermediate in the primary, n = 2 reaction.
A small group of molybdoenzymes, exemplified by DMSOR, are exceptional because these proteins contain no other prosthetic groups in addition to the active-site molybdopterin. Furthermore, substitution of W for Mo in Rhodobacter capsulatus DMSOR affords an active enzyme with an unmodified 3D structure. The Mo(VI) and W(VI) metallopterin UV–vis spectra of this enzyme have been reported to exhibit several broad peaks in the visible and near-UV region with extinction coefficients of the order of ε ≈ 2 mM−1 cm−1 [7], and the molybdopterin absorption has been used as monitor in a pre-steady-state kinetics study [8].
Following the DMSOR, the next step up in optical complexity is found in the AOR family of tungstoenzymes, which all contain—in addition to the tungstopterin active site—a single [4Fe–4S](2+;1+) cluster for electron transfer. The visible spectrum of the iron–sulfur cubane consists of a single broad line at approximately 400–430 nm, which is not only a simpler pattern to that found for [2Fe–2S] clusters (e.g., in the xanthine oxidase family of molybdoenzymes), but it also has a significantly smaller extinction coefficient than flavin or heme prosthetic groups, and so chances are more favorable for monitoring the weaker absorptions from the metallopterin cofactor in these complex enzymes. The homotetrameric (4 × 69 kDa) Pyrococcus furiosus FOR is a member of this family, and we describe here a study of this enzyme as the first example of pre-steady-state kinetics research of a complex, group-6 metalloenzyme using the color of the active center as the monitor. Our choice of FOR was also indicated by the availability of a 3D structure, which was the basis of an initial proposal of part of the enzyme’s working mechanism [6], and was further stimulated by the unusual behavior of the enzyme’s substrate in aqueous solution [9, 10].
Materials and methods
P. furiosus (DSM 3638) was grown at 90 °C under anaerobic conditions with starch as a carbon source, as previously described [11]. Cells were broken by osmotic shock, diluting with 5 vol 30 mM tris(hydroxymethyl)aminomethane (Tris)/HCl, pH 8.0, containing, 0.1 mg ml−1 DNase I, 0.1 mg ml−1 RNase, and 1 mM cysteine. A cell-free extract was obtained as the supernatant after 30 min centrifugation at 15,000g.
“High-activity” FOR was purified as reported previously [9, 10]. Samples were anaerobically purified in 20 mM Tris/HCl buffer, pH 8.0. An extra column of (diethylamino)ethyl (2.0 cm × 10 cm) was equilibrated with 20 mM piperazine/diethanesulfonic acid buffer, pH 6.4. A 100-ml gradient was used from 0 to 0.5 M NaCl. Ferredoxin was purified as reported previously [12]. Samples were anaerobically purified in 20 mM Tris/HCl buffer, pH 8.0. Protein concentration was determined using the bicinchoninic acid method using bovine serum albumin as the standard. Subunit molecular weight and degree of purity were determined with sodium dodecyl sulfate polyacrylamide gel electrophoresis using a Phast system (GE Healthcare).
FOR activity was routinely assayed at 80 °C, under anaerobic conditions, with formaldehyde, or glutardialdehyde, as the substrate and 3 mM benzyl viologen as the electron acceptor in 50 mM 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid (Epps) buffer, pH 8.4.
Steady-state kinetics of FOR were determined by activity measurements in triplicate in an optical assay with different concentrations of formaldehyde (0.5, 1, 2, 5, 10, 25, 50 mM) as a substrate. Benzyl viologen (ε 600 = 10.6 mM−1 cm−1 [13]) or ferredoxin (ε 400 = 17.0 mM−1 cm−1 [14]) were used as an electron acceptor at various concentrations in 50 mM Epps buffer, pH 8.4, and 80 °C, where one unit is defined as 2 μmol electron acceptor reduced per minute. The concentrations of benzyl viologen and ferredoxin were, respectively, 50–3,000 and 10–200 μM, and the ferredoxin concentration is per dimer [15].
The UV–vis spectrum was recorded with a Hewlett-Packard 8452A diode-array spectrophotometer. The optimal excitation wavelength for fluorescence measurements was obtained using a Shimadzu RF-5001PC spectrofluorophotometer. Pre-steady-state studies were carried out under anaerobiosis in 50 mM Epps buffer, pH 8.4, at 50 °C with formaldehyde (25 mM) and formaldehyde-d 2 (25 mM) as a substrate using an SX.18 MV stopped-flow apparatus equipped with a sequential mixing capability and a diode-array rapid-scan detection system (Applied Photophysics). The UV–vis data were obtained by recording 400 spectra at different time scales (16, 65 and 250 s). A spectrum obtained 2 ms after mixing was taken as the “oxidized” reference spectrum. Fluorescence data were obtained by exciting the samples at 280 nm, and the change in maximum fluorescence emission was measured at 340 nm.
Results
Steady-state kinetics
Steady-state kinetics studies at 80 °C were performed on the combinations FOR/formaldehyde/benzyl viologen and FOR/formaldehyde/ferredoxin. FOR catalyzes a two-electron transfer from formaldehyde to the one-electron acceptor benzyl viologen or ferredoxin. This results in a reaction with three substrates; one formaldehyde molecule and two benzyl viologen or two ferredoxin molecules. The possible mechanisms for this reaction are so-called enzyme-substitution mechanisms: the triple-transfer mechanism or the concerted-substitution mechanism [16].
Triple-transfer mechanism:
Concerted-substitution mechanism:
The equation for the reaction rate can be simplified, when substrates B and C (ferredoxin or viologen) are the same. The results have the same form as equations for the reaction rate of an enzyme with two substrates. The triple-transfer mechanism will transform into the substituted-enzyme mechanism for two substrates and the concerted-substitution mechanism will transform into the ternary-complex mechanism for two substrates [16]. The apparent K M values for the second and third substrates are identical, and the measured K M is twice the real K M of the second or third substrate [16, 17]. To predict whether the systems function according to a ternary-complex mechanism or a substituted-enzyme mechanism, one substrate was varied, while the other substrate concentration was held constant.
Substrate inhibition of one of the substrates is described by Eq. 3, where K siB is the constant that defines the strength of the inhibition of substrate B with concentration b:
Primary plots of the concentration of the inhibitor divided by the reaction rate (b/v) versus the concentration of the inhibitor (b) are parabolic and intersect at a common point on the b/v axis. The primary plots of the concentration of the noninhibiting substrate divided by the reaction rate (a/v) versus the concentration of the noninhibiting substrate (a) are linear and intersect at a positive value of the concentration of noninhibiting substrate [16]. A more complex situation occurs when both substrates show substrate inhibition. At low concentrations of both substrates the shapes of both curves are parabolic. Substrate inhibition by one substrate at low concentrations of the other provides strong positive evidence that the substituted-enzyme mechanism applies [16].
The plots of a/v versus a and b/v versus b, with substrate A is formaldehyde and B is benzyl viologen or ferredoxin, are presented in Figs. 1 and 2. All intersection points are situated within experimental error at positive values of the substrates. The parabolic shape of all the figures indicates, as stated above, substrate inhibition by both substrates. All four graphs in Figs. 1 and 2 are consistent with a substrate substituted-enzyme mechanism.

Primary plots for Pyrococcus furiosus formaldehyde oxidoreductase with formaldehyde as a substrate and benzyl viologen as an electron acceptor. Formaldehyde was used in concentrations of 1, 2, 5, 10, 25, and 50 mM. Benzyl viologen was used at concentrations of 50, 100, 500, 1,000, 2,000, and 3,000 μM. Each data point is the average value from three measurements

Primary plots for P. furiosus formaldehyde oxidoreductase with formaldehyde as a an substrate and ferredoxin as electron acceptor. Formaldehyde was used at concentrations of 0.5, 2, 5, 10, 25, and 50 mM. Ferredoxin was used at concentrations of 10, 20, 40, 80, 100, and 200 μM. Each data point is the average value from three measurements
One-substrate Michaelis–Menten kinetics was used to fit the data and to calculate K appM and V appmax at the different concentrations of substrate and acceptor. To calculate the K M and the V max of the different substrates the K appM and v app data were fitted using the equations for the apparent values of the Michaelis–Menten parameters for the substituted-enzyme mechanism [16]:
with equivalent expressions for substrate A.
The K M and V max values of formaldehyde measured with excess benzyl viologen as an electron acceptor (Table 1) are in agreement with previous measurements [10, 18]. The K M value for formaldehyde (3.8 mM) with ferredoxin as an electron acceptor is approximately 3 times less than when benzyl viologen is used as the acceptor. The conversion rate of formaldehyde with ferredoxin as the electron acceptor is 6 times less than when benzyl viologen is used as the acceptor. The values for K M are unusually high for a physiological substrate. We have previously provided experimental evidence indicating that only “free” formaldehyde (i.e., the nonhydrated form which is 0.2% at 80 °C) is a substrate for FOR [10]. The corrected K M values for free formaldehyde with acceptor benzyl viologen or ferredoxin are, respectively, 71 and 21 μM. Note that V max decreases by a factor of 4 when the temperature is lowered from 80 to 20 °C, and that K M for free formaldehyde is temperature-independent [10].
Varying the formaldehyde concentration gives the K M and V max values for the electron acceptors. The K M value of benzyl viologen of 73 μM is comparable to values found in literature for other enzymes that react with benzyl viologen as an electron acceptor. The K M value of ferredoxin (14 μM) is, however, uncharacteristically low: previously reported literature values for FOR and AOR are 100 and 200 μM, respectively [9, 19]. An explanation for this discrepancy may be found in the fact that high concentrations of substrate, up to 50 mM of total formaldehyde for FOR, have been used in previous studies to determine K M of ferredoxin. In the present work concentrations from 0 to 10 mM were used to determine K appM and v app. When activity was measured with high concentrations of formaldehyde (i.e., resulting in significant substrate inhibition) the apparent K M of ferredoxin increased to approach values found in the literature (data not shown).
In a previous study alternative substrates for FOR were reported with K M values of the order of a few millimoles per liter. The lowest K M value was found for glutardialdehyde (K M = 0.8 mM) with benzyl viologen as an electron acceptor [9]. We have been unable to reproduce these results. In our hands the K M for glutardialdehyde was determined to be 12 mM, i.e., approximately the same as the apparent K M for total formaldehyde.
Pre-steady-state kinetics
Pre-steady-state kinetics studies were performed on FOR (20 μM) and formaldehyde at 50 °C. Formaldehyde was used at a concentration of 25 mM (50 μM free formaldehyde). UV–vis spectra were recorded at 40- and at 160-ms intervals. At least two spectra per time point were averaged for noise reduction. In the spectrum of oxidized FOR the broad feature of the [4Fe–4S]2+ cluster dominates. However, close inspection reveals weak (ε max ≈ 1–2 mM−1 cm−1) absorptions at higher wavelengths similar to those reported for the single-cofactor DMSOR [20]. After subtraction of the spectrum at t = 2 ms, difference spectra were obtained (Fig. 3), at 0.1, 0.5, 1, 2, 5, 10, and 16 s, which reveal peak shifts and a lack of isosbestic points.

Difference in absorbance versus wavelength measured in pre-steady-state experiments with formaldehyde as a substrate at times 0.1, 0.5, 1, and 2 s (a) and 2, 5, 10, and 16 s (b). The reference spectrum was taken at t = 2 ms. c UV–vis spectra of the oxidized and the substrate-reduced form (after 65-s incubation time) of P. furiosus formaldehyde oxidoreductase
From the difference spectra five different wavelengths were selected: 335, 390, 435, 595, and 695 nm. At 595 and 695 nm, approximately corresponding absorption bands can be found in the spectrum of oxidized DMSOR from R. capsulatus [8]. Amplitude versus time traces of the 40 and 160 ms spectra at the different wavelengths were combined. After 65 s the time traces for all the wavelengths essentially leveled off (Fig. 4). Data points were taken up to 250 s but no further significant changes were found (not shown).

Stopped-flow UV–vis traces obtained for the reduction of P. furiosus formaldehyde oxidoreductase by formaldehyde at 335, 390, 435, 595, and 695 nm
The major changes at the different wavelengths occur in the first seconds. Three phases starting at different times can be identified in Fig. 5a. The first phase starts at or before t ≈ 20 ms; the second and the third phases start at approximately 100 and 300 ms, respectively. Changes in the amplitude versus time traces at 390 and 435 nm start at or before 20 ms; the trace of the wavelength of 595 nm follows the trend of the 335 nm trace, with the difference that all phases start approximately 200 ms later. All data at the different wavelengths were globally simulated with a four-phase exponential least-squares fit (Table 2).

Stopped-flow UV–vis traces plus fits on a logarithmic time scale obtained for the reduction of P. furiosus formaldehyde oxidoreductase by a formaldehyde or b deuterated formaldehyde at 335, 390, 435, 595, and 695 nm. c Stopped-flow fluorescence trace plus fit obtained for the reduction of P. furiosus formaldehyde oxidoreductase by formaldehyde at an excitation wavelength of 295 nm and an emission wavelength of 340 nm. d Stopped-flow UV–vis traces plus fits obtained for the binding of formate by P. furiosus formaldehyde oxidoreductase at 335, 390, 435, 595, and 695 nm
A previous steady-state kinetics study on the substrate kinetic isotope effect of FOR showed a significant primary isotope effect for C–H/D bond breaking. The specific activity of FOR dropped by a factor of 3 when deuterated formaldehyde was used as a substrate [21]. In the present work the pre-steady-state kinetics of FOR with formaldehyde-d 2 was determined in the same fashion as for the experiments with formaldehyde. The amplitude versus time traces at selected wavelengths of FOR incubated with formaldehyde-d 2 resemble the traces of the different wavelengths when formaldehyde was used as the substrate; the phases are similar, only the duration and the onset of the changes of states are different. Again the data were fitted with a four-phase exponential fit (Fig. 5b; Table 2).
Tryptophan residues are excellent probes for protein conformational changes. In FOR a single tryptophan (Trp441) can be found in the active site [6]. A fluorescence stopped-flow experiment was performed using 280 nm as the excitation wavelength and 340 nm as the emission wavelength. The concentrations of FOR and formaldehyde used were the same as in the previous UV–vis stopped-flow experiments. Traces were measured on different time scales: 1, 10, and 100 s, and were combined (Fig. 5c). The data can be fitted to a three-phase exponential model. The noise levels however are high. The fluorescence data were fitted simultaneously with the UV–vis data of Fig. 5a, affording a consistent set of parameters (Table 2).
Formaldehyde is oxidized to formate by FOR with reduction of the W(VI) center. We checked whether FOR can oxidize formate to carbon dioxide. Activity measurements in an optical assay were done at 80 °C using formate as the substrate and methyl viologen as the electron acceptor. FOR was unable to oxidize formate. Pre-steady-state binding experiments with oxidized FOR using formate as the nonreducible substrate were done at the same time intervals as for the experiments with formaldehyde (Fig. 5d). Although formate is not converted to carbon dioxide by FOR, absorption changes are found in the time traces at the different wavelengths. The first phase starts at t ≈ 20 ms; the second and the third phases start at approximately 100 and 300 ms, respectively. The time trace at 335 nm resembles the trace obtained with formaldehyde. The time traces at 390 and 435 nm are not similar to each other in contrast to when formaldehyde was used as the substrate. The time traces at 595 and 695 nm are essentially unaffected by the formate. All data at the different wavelengths were globally fitted with a two-phase exponential least-squares model (Table 2).
Discussion
Steady-state kinetics
An initial proposal for part of the working mechanism of P. furiosus FOR was deduced by Hu et al. [6] from the enzyme’s 3D structure: two electrons are transferred from the substrate to the tungsten center. The pterin acts as a noninnocent ligand and transfers the electrons via the N8 atom one by one through Cys491 to the iron–sulfur cluster of FOR. The iron–sulfur cluster of ferredoxin accepts an electron through its Asp14 [6]. The electron is then transferred from the iron–sulfur cluster of ferredoxin to a membrane-bound hydrogenase [22]. After reduction of the iron–sulfur cluster of ferredoxin, the electron transfer comes to a halt. Only one electron at a time can pass through this route. Two ferredoxin molecules are necessary to transport both electrons; alternatively, a ferredoxin dimer [15] can twice in a row bind to FOR and accept an electron. This means that a two-substrate mechanism is insufficient to describe this three-substrate system. The second and the third substrates are both ferredoxin. This results in steady-state Michaelis–Menten equations that have the same form as equations for the reaction rate of an enzyme with two substrates; however, the measured K M for ferredoxin will be twice the “real” K M.
The triple-transfer mechanism for three substrates with substrates two and three (ferredoxin) the same is consistent with the experiments.
Both substrates, either formaldehyde and ferredoxin or formaldehyde and benzyl viologen, inhibit FOR. The K M values of 20–70 μM for free formaldehyde are in agreement with our previous studies [10]. When a nonphysiological electron acceptor such as benzyl viologen is used then the K M for formaldehyde is 3–4 times higher. It is possible that when ferredoxin binds to FOR the enzyme experiences a slight conformational change, resulting in a lower K M for formaldehyde.
In these studies a K M of 14 μM was determined for ferredoxin, which is 5 times lower than the K M of benzyl viologen. The high K M reported in the literature for ferredoxin was determined with high concentrations of formaldehyde. An explanation for this high value is that at high concentrations (50 mM) formaldehyde inhibits and partially denatures FOR.
Pre-steady-state kinetics
Pre-steady-state kinetics studies were performed at 50 °C. This relatively low temperature was dictated by technical limitations of the stopped-flow apparatus. FOR activity at 50 °C is 2.8 times less than at 80 °C, which affords an increased time resolution of enzyme intermediates.
Difference spectra from the pre-steady-state data revealed peak shifts and a lack of isosbestic points. These features are an indication that several processes are happening in the first seconds of the reaction. The visible spectrum of the iron–sulfur cubane consists of a single broad line at approximately 400–430 nm; the relatively weakly colored metallopterin is found at different wavelengths.
A minimal first-order exponential fit model was used to fit the pre-steady-state data. The UV–vis data could be fitted globally with a three-phase exponential fit. Two fast processes and one slow process were found. The fluorescence data were initially modeled with a two-phase exponential fit, resulting in one fast and one slow process. The fast and slow processes deduced from the fluorescence and from the UV–vis data were clearly different. A four-phase exponential model afforded a significantly improved fit to the UV-data (two fast and two slow processes), and the fluorescence data were fitted more accurately with a three-phase exponential model (two fast processes and one slow process). The complete set of fluorescence and UV–vis data obtained with the same enzyme and substrate were then fitted in a global analysis with a three-phase and a four-phase exponential model, respectively. The same four-phase model was used to fit the pre-steady-state data obtained using formaldehyde-d 2 as the substrate. The fit is comparable with the fit of formaldehyde but the rate constants of the first and second phases are lower, as expected from the steady-state studies. The first phase is 4 times slower (k 1 = 1.12 s−1) than with formaldehyde as the substrate. The rate constant of the second phase (k 2 = 0.94 s−1) is lower than when formaldehyde is used as the substrate, consistent with a previously reported steady-state kinetics experiment [21]. The rates for the first two phases with CD2O as the substrate are not significantly different within experimental error. The rate constant for intramolecular electron shuffling appears to be unaffected by the use of formaldehyde-d 2.
Residuals of the individual fits plotted versus time were essentially randomly distributed around the x-axis, thus attesting to the quality of the global fit. Several intermediate species are formed during the first seconds of the reaction. These results are in contrast with previous studies on molybdenum–DMSOR, where in stopped-flow experiments clear isosbestic points were found, and analysis of the time course produced a simple exponential, indicating a simple concerted two-electron reduction of the enzyme with no intermediate species [23].
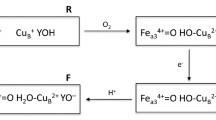
Two alternative interpretations can be envisioned for the three phases in pre-steady-state kinetics (Fig. 6).

Proposed alternative models for the pre-steady-state phases of fully oxidized P. furiosus formaldehyde oxidoreductase reacting with formaldehyde. Rate constants with capital letters as a subscript involve binding, oxidizing the substrate, releasing the product, and rearranging electrons between the tungsten center and the iron–sulfur cluster. Alternatively, rate constants with numbered subscripts involve oxidation of substrate, a conformational change leading to weakened product binding, release of product, and electron rearrangement
FOR is represented in Fig. 6 as a rectangle with two redox groups, a tungstopterin (W) with oxidation states 6+, 5+, or 4+, and a [4Fe–4S] cubane cluster with oxidation states 2+ or 1+. On the left of W is an empty binding pocket which can be occupied by substrate (A), by product (P), or by ready-to-leave deactivated product (P*). Occupation by A means that the enzyme has acquired a molecule of substrate in the previous reaction. Similarly, a W capped by a double-bonded O means that the enzyme has previously acquired an oxygen (protonation undefined) from the solvent. The protein is initially in the fully oxidized form; the tungsten center is 6+ and has an oxygen atom bound that was previously acquired from a water molecule, and the iron–sulfur cluster is 2+.
In the first model the initial step (k A = 4.7 s−1) observed in the optical monitoring is taken to be the overall binding process of formaldehyde to the enzyme. No changes to the formal oxidation state of the tungsten center and iron–sulfur cluster are assumed to occur in this stage. Subsequently, in what appears to be a single event, the oxygen atom is transferred, the substrate is oxidized to the product (k B = 1.9 s−1), and the tungsten center receives two electrons from the substrate and is reduced to 4+. The product then leaves the protein (k C = 6.10 × 10−2 s−1). In this step there are no changes in the oxidation states of the tungsten center and the iron–sulfur cluster. The active site of FOR has been proposed by Hu et al. [6] to be closed off from the solvent by a lid provided by the protein itself. It is possible that the substrate, free formaldehyde, is so small that it will not experience significant resistance in reaching the active site. The bigger and charged product may have more difficulty getting free of the enzyme, resulting in a low rate. The protein will then, in the absence of an external electron acceptor, redistribute (to a minor extent; see below) its electrons over the tungsten center and the iron–sulfur cluster (k D = 2.18 × 10−2 s−1). The tungsten center will become 5+ and the iron–sulfur cluster 1+, consistent with previously determined redox properties for “high-activity” enzyme [10]. An isotope effect of 4.2 is found for the rate constant of the first step (Table 1), which seems to be unrealistically large for a noncovalent binding event, and would rather seem to point toward an event involving the breaking and making of covalent chemical bonds. Therefore, the following would appear to be a rather more likely interpretation of the stopped-flow data.
Alternatively, if the entry and binding of the substrate does not lead to a detectable change in the optical absorption, then the first step (k 1 = 4.7 s−1) can be the two-electron reduction of the tungsten. In this model the second step (k 2 = 1.9 s−1) is a conformational change leading to an “open” state with decreased affinity for the product formed. The third step (k 3 = 6.10 × 10−2 s−1) then is the ready release of this now weakly bound formate. The pre-steady-state phase ends with the two-electron-reduced intermediate which, in the absence of an external electron acceptor, slowly, and only partially converts, by electron shuffling, to the EPR-detectable, paramagnetic side product. Previous EPR studies on FOR revealed this slow electron shuffling at 20–60 °C between the tungsten center and the iron–sulfur cluster upon reduction with formaldehyde or with dithionite [10]. Only after minutes the W(V) signal appeared in the EPR measurements. This observation is in qualitative agreement with the low rate constants found here for the decay of the fourth state.
The pre-steady-state data from the experiments with formate and FOR were fitted with a two-phase exponential model: there is one fast process and one slow process. Formate is not oxidized or reduced by FOR; therefore, the fast process (k obs1 = 0.51 s−1) could be the entry and the binding of formate to the active site of FOR. As in the FOR/formaldehyde system the slow process (k obs2 = 2.00 × 10−2 s−1) is probably a nonphysiological process: the protein reaches a dead end.
Excluding the very slow electron redistribution step in the absence of external electron acceptor (the last step in Fig. 6) a complete cycle could now be written in which the enzyme shuttles between fully oxidized, one-electron-reduced, and two-electron-reduced, by inclusion of the reoxidation steps of the two-electron-reduced intermediate through serial reactions with two molecules of oxidized ferredoxin. However, such a scheme would be inconsistent with steady-state turnover rates, because the rate constant (k 3 or k C) of the product-release step (the penultimate step in Fig. 6) is more than an order of magnitude less than k cat. On the other hand, it is conceivable, that the one-electron-reduced intermediate reacts with a second substrate molecule to become three-electron-reduced (with respect to the resting enzyme). This would lead to the cycle presented in Fig. 7 in which the enzyme shuttles between one-electron-reduced, two-electron-reduced, and three-electron-reduced.

Proposed model for the catalytic redox cycle of P. furiosus formaldehyde oxidoreductase. A is the substrate formaldehyde; P is the product formate. The cube-enclosing circle represents the natural redox partner, ferredoxin. Pre-steady-state kinetics measures either the steps with rate constants k A through k D or, alternatively, those with constants k 1 through k 4. The fully oxidized enzyme is reductively activated by the reaction with formaldehyde to enter a redox cycle in which the enzyme shuttles between one--electron-reduced, two--electron-reduced, and three-electron-reduced states. See the text for additional explanation
Under this model the steps observed in the pre-steady-state experiments would actually constitute an activation process. Note that once the steady state of this cycle has been reached, the enzyme reacts sequentially with one formaldehyde and two ferredoxin molecules consistent with the “A+2B” Michaelis–Menten steady-state analysis.
The proposed reaction cycle of Fig. 7 is based on a combination of steady state-kinetics, pre-steady-state kinetics, and EPR studies in the absence of an electron acceptor and available structural data. Future pre-steady-state kinetics studies in the presence of excess natural electron acceptor ferredoxin may put the model to a rigorous, be it experimentally challenging, test.
Abbreviations
- AOR:
-
Aldehyde oxidoreductase
- DMSOR:
-
Dimethyl sulfoxide reductase
- Epps:
-
4-(2-Hydroxyethyl)-1-piperazinepropanesulfonic acid
- EPR:
-
Electron paramagnetic resonance
- FOR:
-
Formaldehyde oxidoreductase
- Tris:
-
Tris(hydroxymethyl)aminomethane
References
Hille R (1996) Chem Rev 96:2757–2816
Johnson MK, Rees DC, Adams MWW (1996) Chem Rev 96:2817–2839
Hille R. (2005) Arch Biochem Biophys 433:107–116
Hille R (2002) Trends Biochem Sci 27:360–367
Chan MK, Mukund S, Kletzin A, Adams MWW, Rees DC (1995) Science 267:1463–1469
Hu Y, Faham S, Roy R, Adams MWW, Rees DC (1999) J Mol Biol 286:899–914
Stewart LJ, Bailey S, Bennet B, Charnock JM, Garner CD, McAlpine AS (2000) J Mol Biol 299:593–600
Adams B, Smith AT, Bailey S, McEwan AG, Bray RC (1999) Biochemistry 38:8501–8511
Roy R, Mukund S, Shut GJ, Dunn DM, Weiss R, Adams MWW (1999) J Bacteriol 181:1171–1180
Bol E, Bevers LE, Hagedoorn P-L, Hagen WR (2006) J Biol Inorg Chem 11:999–1006
Arendsen AF, Veenhuizen PTM, Hagen WR (1995) FEBS Lett 368:117–121
Kim C, Brereton PS, Verhagen MFJM, Adams MWW (2001) Methods Enzymol 334:30–40
Corbin JL, Watt GD (1990) Anal Biochem 186:86–89
Zhou ZH, Adams MWW (1997) Biochemistry 36:10892–10900
Hasan MN., Hagedoorn P-L., Hagen WR (2002) FEBS Lett 531:335–338
Cornish-Bowden A (1995) Fundamentals of enzyme kinetics. Portland, Cambridge
Dalziel K (1968) Biochem J 114:547–556
Dhawan IK, Roy R, Koehler BP, Mukund S, Adams MWW, Johnson MK (2000) J Biol Inorg Chem 5:313–327
Roy R, Menon AL, Adams MWW (2001) Methods Enzymol 331:132–144
Stewart LJ, Bailey S, Bennett B, Charnock JM, Garner CD, McAlpine AS (2000) J Mol Biol 299:593–600
Hagedoorn P-L (2002) PhD thesis, Delft University of Technology. http://repository.tudelft.nl/file/80698/007489
Silva PJ, Van den Ban ECD, Wassink H, Haaker H, De Castro B, Robb FT, Hagen WR (2000) Eur J Biochem 267:6541–6551
Adams B, Smith AT, Bailey S, McEwan AG, Bray RC (1999) Biochemistry 38:8501–8511
Acknowledgement
This research was supported by a grant from the Council for Chemical Sciences of the Netherlands Organization for Scientific Research (700.51.301).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bol, E., Broers, N.J. & Hagen, W.R. A steady-state and pre-steady-state kinetics study of the tungstoenzyme formaldehyde ferredoxin oxidoreductase from Pyrococcus furiosus . J Biol Inorg Chem 13, 75–84 (2008). https://doi.org/10.1007/s00775-007-0301-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-007-0301-3