
Abstract
Battery fast charging is pivotal for broader acceptance of electric mobility. While demonstrated for lithium titanate \((\text {Li}_4\text {Ti}_5\text {O}_{12},\) LTO) anodes, the underlying mechanisms are still poorly understood. Recently, NMR \(T_1\) relaxation time constants of \({}^7\text {Li}\) in the bulk of LTO were found to change if the surrounding electrolyte was altered. It was explained by interdiffusion of mobile lithium ions between the two phases, facilitated by unpinning of polarons from surface defects and leading to a pseudocapacitive effect that potentially influences fast charging. This effect is explored further by systematically varying the lithium salt concentration in an aprotic electrolyte in contact with LTO. Spectrally resolved \({}^7\text {Li}\) \(T_1\) NMR relaxation times were used as a measure for bulk concentration changes of paramagnetic polaronic charges in LTO. Correlation of electrolyte concentration and \({}^7\text {Li}\) \(T_1\) showed qualitatively different behavior above and below a salt concentration of about 5 mM, leading to a relaxation dispersion maximum in the LTO bulk. At intermediate concentrations, relaxation was consistent with a \({}^7\text {Li}\) exchange equilibrium between LTO and electrolyte. Upon contact of the two phases, yet without insertion into an electrochemical cell or applying an external potential, lithium ions redistributed between LTO bulk and liquid electrolyte. The results can be understood analogously to the distribution of mobile lithium ions between two phases separated by a \(\text {Li}^{+}\) permeable membrane. This is the first demonstration of such an equilibrium for non-faradaic lithium exchange at an interface between a solid ceramic electrode and a liquid electrolyte outside an electrochemical cell, substantiating our previous hypothesis of a polaron-supported mechanism. This study provides a basis for more quantitative (surface)-defect engineering, which is key to optimize battery fast-charging properties.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Driven by consumer demand, the development of battery materials capable of sustaining fast charging is a major focus of current research [1]. Fast charging represents a boundary between battery- and capacitor-type charging mechanisms [2, 3]. The energy density of batteries and the power density of capacitors are combined in supercapacitors to reinforce the positive features of both principles [4, 5].
Materials for supercapacitors are often carbon based. Balancing of energy and power density may be achieved by further employing either electrodes from hybrid materials, or asymmetric cells containing one electrode with dominantly non-faradaic or pseudocapacitive charge storage and one electrode that consists of a lithium intercalation material [6]. Nanostructured lithium titanate \((\text {Li}_4\text {Ti}_5\text {O}_{12};\) LTO) has been suggested as possible intercalation material for supercapacitors due to its excellent cycling stability even at high C-rates [7]. Also, despite a low lithium ion mobility and low electronic conductivity in the stoichiometric form, variations of nano-LTO have been synthesized that can be cycled at rates of several hundred C [8].
A major challenge is posed by the optimization of synthesis conditions to obtain materials with the desired properties. It is not even well understood which analytical parameters to employ as targets for material optimization. Only few analytical techniques are available for a detailed analysis of the nature of these defects [9,10,11]. Nuclear magnetic resonance (NMR) has long been used to investigate the dynamics in multi-phase systems [12, 13]. These techniques can also be adopted for battery materials.
For LTO anode material, a correlation was proposed between vacancies on electrolyte accessible surfaces and Li-ion mobility in the bulk of the material, induced purely by contact of LTO with Li ion containing electrolytes. Thereby, intercalative pseudocapacitance and fast-charging capability may be facilitated [14]. Such a relationship was inferred from the spectrally resolved longitudinal, or \(T_1,\) NMR relaxation behavior of \({}^7\text {Li}\) in LTO. When brought in contact with a liquid electrolyte, a \(T_1\) change was observed not only in the electrolyte [15] or on the directly affected surface, but also in the bulk of the ceramic material. Lithium ions entered the defect-containing surface of the material, leading to the unpinning of paramagnetic polaronic charges associated with these defects.
Polarons are mobile quasiparticles with negative charge that are neither fully localized nor located in the conduction band of a solid material. Mobility is influenced by dielectric properties and phonon oscillations of the material [9]. Polaron mobility can be associated with an effective mass that differs from the electron mass, and depending on the dimension of charge delocalization in relation to the lattice constant of a material, large and small polarons are distinguished [16]. For the experimental characterization of battery materials, the polaron concept is only occasionally employed [17], while theoretical studies have endorsed the concept for some time [18]. More recently, properties of LTO have been discussed theoretically in the context of polarons [19,20,21]. Experimentally, the absence of a room temperature electron paramagnetic resonance (EPR) signal in reduced or aliovalently doped LTO despite the nominal presence of paramagnetic \(\text {Ti}^{3+}\) centers indicates the suitability of the polaron concept for this material, particularly since freezing out of the electron mobility is possible and the EPR spectrum becomes visible at low temperatures [10].
Here, it is investigated if there is a systematic correlation between lithium concentration in the electrolyte and the observed \({}^7\text {Li}\) \(T_1\) relaxation time constant in the solid using a concentration series of the electrolyte salt. The observed results are compared with theoretical predictions, and a model is devised for polaronic electron mobility in stoichiometric nano-LTO. For intermediate electrolyte concentrations, the suitability and limitations of a simple two-phase equilibrium model for lithium redistribution are discussed.
2 Sample and Experiment Details
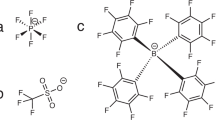
All preparation steps were done in a glove box with argon atmosphere. For each experiment, a Young-type NMR tube was filled with 100 mg of nano-LTO powder from Sigma-Aldrich, with a manufacturer-specified particle size below 200 nm and a specific surface area of 32.6 \({{\text {m}^2/\text {g}}}.\) Then, 1.5 mL of electrolyte was added, which consisted of dimethylcarbonate (DMC) and lithium bis(trifluoromethanesulfonyl)imide \((\text {LiN}(\text {SO}_2\text {CF}_3)_2;\) Li-TFSI), see Fig. 1. The concentrations of the prepared electrolytes were 0.13 mmol/L, 0.6 mmol/L, 2.7 mmol/L, 12.2 mmol/L, 54.6 mmol/L and 250.0 mmol/L. The tube was shaken until a homogeneous slurry was obtained, and then left undisturbed during a settlement period of 24 h to ensure that the sample was in equilibrium state during NMR measurements. As shown schematically in Fig. 1a, a dense slurry was formed at the bottom of the tube, with excess electrolyte as clear supernatant.

Illustration of the LTO/electrolyte sample and the data analysis procedure. a Schematic of the sample after settling. Relaxation measurements were conducted by placing the suspension in the sensitive volume of the NMR coil. b Spectrally resolved \({}^7\text {Li}\) \(T_1\) relaxation time distribution of the LTO sample in contact with 250 mmol/L Li-TFSI in DMC electrolyte. The \({}^7\text {Li}\) NMR spectrum consists of a broad signal from Li in LTO and a narrow resonance from Li in the electrolyte. The spectrum was referenced to the electrolyte resonance at 0 ppm. To emphasize weak signal contributions, the pseudocolor plot was scaled with the square root of the amplitude while maintaining the sign of the distribution. At the top, the \({}^7\text {Li}\) NMR spectrum obtained with the longest recovery delay is shown. The y-axis shows the signal-to-noise ratio (SNR). The magenta, blue, and red bars represent the spectral regions that were integrated to obtain the 1D distributions to the right of the 2D plot. The three distributions showed a similar amplitude without additional scaling. The negative contributions in the distribution were caused by Li exchange between the electrolyte and LTO. c Residuals between data and fit, also in units of SNR. No systematic features are apparent that would indicate signal contributions not consistent with the chosen exponential kernel. The black line represents the spectrally resolved standard deviation of the residuals. Since the data were scaled to unity variance before conducting the Laplace inversion, the standard deviation should be equal to one across the whole spectrum (color figure online)
With the tube placed in the probe such that the sensitive volume of the NMR coil contained only the slurry, static \({}^7\text {Li}\) saturation recovery \(T_1\) relaxation measurements were performed on a Bruker Avance III HD 600 wide-bore spectrometer equipped with a gradient probe (Bruker PA BBO 600W2/S4 BB-H &F-D-05 DIFF). The number of accumulations was 8, using \(\pi /2\) pulses of \(15\,\upmu \text {s}\) duration at 50 W radio frequency (rf) power. A series of up to 35 delays was used, logarithmically spaced between \(10^{-5}\) s and 500 s, to determine the buildup of the \({}^7\text {Li}\) signal. Afterwards the supernatant electrolyte was decanted and transferred into another NMR tube. \({}^{19}\text {F}\) spectra were acquired on the same instrument, accumulating 16 scans, using \(\pi /2\) pulses of \(23\,\upmu \text {s}\) at 27.6 W rf power. To determine the relative change of the electrolyte salt concentration in the supernatant of the electrolyte/LTO suspension, an equal amount of the supernatant and of the neat electrolyte were filled into separate NMR tubes and the area of the \({}^{19}\text {F}\) signal compared. The relative change was calculated by dividing the areas.
Laplace inversion was performed on two-dimensional, spectrally resolved relaxation data using uniform penalty regularization [22], which is a Tikhonov regularization algorithm in generalized form. Regularization was also conducted along the spectral, not inverted dimension [23]. A zero-crossing penalty instead of a non-negativity constraint was used to minimize sign changes in the calculated distribution that are not supported by the data [24]. Analyses were done with GNU Octave (version 4.0.3) [25] using a home-written function for Laplace inversion. The regularization parameters have been chosen as detailed in [26]. The \(T_1\) distribution was calculated with 100 logarithmically spaced time constant values between \(10^{-2}\) and \(10^4\) s. Thereby, it was possible to separate relaxation processes in solid lithium conducting materials [27, 28]. Since the signal relaxes towards a maximum in saturation recovery experiments, the signal value at infinite time was fitted as a single data point for each frequency rather than as part of the distribution at very long time constants [22].
3 Results
A series of colloidal suspensions was prepared, consisting of nano-LTO powder in DMC electrolyte containing six different Li-TFSI concentrations, ranging from 0.1 mmol/L to 250 mmol/L. Figure 2 shows the \(T_1\) relaxation time distribution of \({}^7\text {Li}\) in the LTO solid of the different samples. It was obtained by calculating a spectrally resolved \(T_1\) distribution, as shown in Fig. 1b, and then integrating this distribution in the spectral region downfield and, independently, upfield of the electrolyte resonance of the \({}^7\text {Li}\) NMR spectrum. The projections do not include the resonance from \({}^7\text {Li}\) in the electrolyte; thus, the relaxation mode caused by the solvent at \(T_1\) of about 1 s is not visible. This analysis protocol provided results that were consistent with, yet more robust than summing the spectra for the different recovery delays and then performing a Laplace inversion of a one-dimensional data set. In addition, three previously published data sets of the pure solvent, of 1 M and of 2 M \(\text {LiPF}_{6},\) recorded with the same parameters but recovery delays only up to 400 s, were used to extend the range of concentrations [14]. As was shown previously, the effect of varying the anion from Li-TFSI to \(\text {LiPF}_{6}\) on \(T_1\) of \({}^7\text {Li}\) in LTO is marginal.

\(T_1\) relaxation time distributions of \({}^7\text {Li}\) in LTO for different electrolyte concentrations. To exclude the contribution from \({}^7\text {Li}\) in the electrolyte itself, the distributions were obtained by summing the spectrally resolved \(T_1\) distributions downfield (a, c) and upfield (b) of the electrolyte resonance. d Signal decay towards equilibrium vs. recovery time for the downfield contribution of the resonance. The legend in a represents electrolyte concentrations in mmol/L
With a particle size of about 200 nm, the main contribution of the broad resonance in the \({}^7\text {Li}\) NMR spectrum (Fig. 1b) originated from Li in the bulk rather than the surface of LTO. Therefore, the obtained \(T_1\) values represent primarily bulk properties. However, the surface area was large enough that surface species with a different relaxation behavior should be distinguishable.
At low concentrations, the \(T_1\) distribution was monomodal, i.e., it showed a single, symmetric feature whose position was dependent on the electrolyte concentration. Therefore, the large majority of Li ions in LTO experienced the same averaged relaxation behavior. If only the surface of the LTO would be affected by the electrolyte, a distribution consisting of multiple relaxation components characteristic for a core–shell configuration would be expected. A gradient from the surface to the interior would cause an asymmetric broadening of the distribution, which was also not observed. The relaxation mode shifted as a function of concentrations, but did not broaden.
At 250 mM, the distribution started to broaden and become asymmetric, indicating the emergence of a distinguishable second component or a distribution of relaxation properties from the surface to the core. This became more apparent as concentrations were increased further, where two different yet still overlapping contributions started to emerge. This is also visible more directly when looking at the relaxation curves whose signal value at infinite time was subtracted, shown in Fig. 2d. At high concentration, the initial signal decay still got faster, yet the tail started to relax more slowly. While a still increasing fast relaxation component may become too fast for effective averaging if motional averaging would be the reason for the monomodal distribution at low concentration, the observation that the slowly relaxing wing of the relaxation distribution at high concentration increases again suggests an emerging spatial dependence of relaxation.
Since nominally stoichiometric, diamagnetic LTO was used, Li mobility would not be sufficient to average out \(T_1\) globally [29]. For overlithiated LTO, a decrease of \(T_1\) is well known [30, 31]. However, the samples investigated here were suspensions that had not been chemically or electrochemically overlithiated, and they had never been inserted into an electrochemical cell. Furthermore, the electrolyte solvent did not lead to swelling of the LTO. Nevertheless, the results showed that just bringing LTO in contact with an electrolyte solution containing a lithium salt sufficed to globally alter \(T_1\) in the bulk of the solid phase.
Li exchange between the electrolyte and the LTO solid was responsible for features with negative amplitudes in the distributions, which are visible more clearly in the relaxation distribution of spectral components upfield of the electrolyte resonance (Fig. 2b). Residuals for the Laplace inversion compatible with random white noise, as shown in Fig. 1c, could only be obtained by allowing for negative components in the distribution. The possible existence of such negative contributions has been justified theoretically for samples with exchanging species [32]. However, it cannot be excluded either that the chosen exponential kernel for the Laplace inversion may not be compatible with the exchange affected signal decay. To distinguish, exchange processes would have to be measured more directly [33].
The positions of the relaxation distribution maxima, shown in Fig. 3, were used for further analysis.

\({}^7\text {Li}\) \(T_1\) relaxation time distribution maxima in LTO during contact with electrolytes at different concentrations. (Black) Maximum of \(T_1\) relaxation time distribution of the whole signal, but without the electrolyte contribution. (Green) \(T_1\) maximum of the signal downfield of the electrolyte resonance. (Orange) \(T_1\) maximum of the signal upfield of the electrolyte resonance. (Purple) Relative change of Li-TFSI concentration in the protruding solution after contact with LTO, determined using \({}^{19}\text {F}\) NMR. The dashed lines show the result of fitting the \(T_1\) values at intermediate concentrations (color figure online)
A finer scale was obtained by interpolating the data points from Laplace inversion with a cubic spline. Without lithium salt in the electrolyte, \(T_1 = 71\) s was determined. Upon increasing the electrolyte concentration to 2 mmol/L, \(T_1\) increased to about 78 s. A further increase of the electrolyte concentration caused a decrease of \(T_1\) to a value of 9 s in a 2000 mmol/L solution. When comparing \(T_1\) in spectral regions downfield and upfield of the electrolyte resonance, different results were obtained below concentrations of 10 mmol/L. At intermediate concentrations below 250 mmol/L, the relaxation behavior was more uniform as a function of \({}^7\text {Li}\) NMR frequency; while at higher frequencies, the difference increased again.
Also shown in Fig. 3 is the electrolyte salt concentration change in the supernatant of the electrolyte/LTO suspension relative to the neat electrolyte by calculating the ratio of the \({}^{19}\text {F}\) NMR signals. In the range of 0.1–10 mmol/L, the supernatant was substantially depleted of electrolyte salt compared to the neat solution. It is indicative of \({}^{19}\text {F}\)-containing electrolyte anion adsorption on the LTO surface. The relative adsorption decreased linearly with the logarithm of concentration, which indicates a saturation of strongly adsorbed anions. At higher concentrations, the salt or ions adsorbed on the LTO surface only marginally changed the electrolyte concentration in the supernatant. At 250 mmol/L, the effect may even be reversed, yet the relative change was on the order of the estimated uncertainty of 2% of this experiment. Nevertheless, a similar effect had been reported in an experiment where the volume of added electrolyte was changed, and the salt concentrations were varied independently [34]. It was suggested that at low amounts of electrolyte, the saturation of small pores in the solid with electrolyte would cause the observed concentration variation, and that as the electrolyte concentration was increased, ion exchange would play a more important role. In the present experiment, where a surplus of electrolyte was available compared to the total pore volume of LTO, the results suggested the existence of an equilibrium between anions adsorbed on the LTO surface and in the electrolyte. As the electrolyte salt concentration was increased, the LTO surface got progressively more covered with anions from the electrolyte; hence, less sites on the LTO surface that allow a strong electrolyte adsorption were available.
The point where the electrolyte salt concentration difference assumed zero coincided with the point where the difference of the \(T_1\) relaxation between upfield and downfield spectral regions disappeared. This indicates that the range between 5 and 10 mmol/L may correspond to a transition region of a multi-step equilibrium [35], although the experimental data did not provide direct evidence regarding the exact nature of the involved equilibria.
4 Discussion
Little information is available regarding the surface structure of polycrystalline ceramic nanoparticles such as the investigated nano-LTO. Rather than a well-defined crystalline surface, a surface complexion, as outlined in Fig. 4, is expected that includes defects to minimize the energy of the surface towards its environment and that may incorporate \(\text {Li}^{+}\) and adsorbed \(\text {TFSI}^{-}\) from the electrolyte [36]. Possible defects may be neutral \(\text {Li}_2\text {O}\) vacancies, or the expulsion of two LiO radicals to form \(\text {Li}_2\text {O}_2\) is also conceivable. Furthermore, LTO synthesis under reducing atmosphere leads to oxygen-deficient material with a blue color [23]. The investigated stoichiometric LTO was white, but the surface contained intrinsic defects from interruption of the lattice periodicity. Moreover, additional Li ions that are charge compensated by a reduction of diamagnetic \(\text {Ti}^{4+}\) to paramagnetic \(\text {Ti}^{3+}\) are possible as well, facilitated by a small excess of Li during synthesis to compensate for Li loss during heat treatment [10] and the ‘zero-strain’ behavior of LTO upon intercalation of additional lithium [37]. The presence of excess Li in LTO has been suggested by the reported increase of \({}^7\text {Li}\) \(T_1\) in bulk LTO if an electrolyte containing a sodium salt is used [14]. Sodium cannot intercalate into LTO, but it may charge compensate the release of Li into the electrolyte at the LTO surface. However, no specific information regarding the surface chemistry can be gathered directly from the presented relaxation data of a static sample.
A simplified thermodynamic model is outlined in Fig. 4. Electrolyte and LTO are not chemically reactive towards each other. In the absence of an external potential, no solid–electrolyte interphase (SEI) forms; hence, the equilibration effects are purely physical.

Schematic depiction of the LTO/electrolyte sample in equilibrium, including the relevant diffusion and redox processes. The red curves indicate qualitatively the chemical potential \(\mu _{{{\textrm{Li}}^{+}}}\) for different electrolyte concentrations (color figure online)
The LTO surface acts as a semi-permeable membrane for \(\text {Li}^{+}\) ions. No other ionic or non-ionic species present in the sample can penetrate the surface. At the same time, if we rule out anionic redox reactions that have been described to occur in cathodes of electrochemical cells [38], the titanium ions are formally the only redox-active species, supporting transitions between \(\text {Ti}^{3+}\) and \(\text {Ti}^{4+}.\) Therefore, for simplicity it is assumed that redox activity is limited to \(\text {Ti}^{3+}/\text {Ti}^{4+}\) processes, which are restricted exclusively to the solid LTO material but distinguished between the LTO bulk and its surface complexion. Exchange is possible between the two environments, accompanied by a transfer of a \(\text {Li}^{+}\) ion,
Subscripts refer to the environment, with (bulk) representing the crystalline environment in the core of an LTO particle and (cx) the surface complexion. This mobility of non-conduction electrons in combination with the possible delocalization of charge between different Ti centers is the primary reason why the polaron concept is suitable for the description of dynamic processes in LTO. To maintain local charge neutrality, the polaron and \(\text {Li}^{+}\) mobility may be correlated [17], but for a qualitative thermodynamic discussion this is disregarded. In the LTO bulk, it is assumed that every \(\text {Ti}_{({\textrm{bulk}})}^{3+}\) is charge compensating a surplus \(\text {Li}^{+},\) while in the complexion different types of defects may occur. As long as they are immobile, they are of lower direct importance for the measured \({}^7\text {Li}\) \(T_1\) relaxation time constants.
In equilibrium, the electrochemical potential \(\tilde{\mu }_{{{\textrm{Li}}}^{+}}\) of lithium ions is equal in the solid and in the liquid phase. For a semi-permeable membrane between two liquids or, more generally, soft matter reservoirs, this is described by the Donnan equilibrium, leading to osmosis [39]. However, while the conceptual principle still holds, the details are somewhat different here. \(\text {Li}^{+}\) generally has a different standard potential in the liquid electrolyte and in the solid phase, and rather than having multiple mobile ionic species, a redox capable immobile transition metal ion is present in the solid phase. Nonetheless, an electrochemical equilibrium may be established, provided that local charge neutrality can be maintained in both phases. For this, three situations have to be distinguished, depending on the electrolyte concentration. Let us first consider a pure electrolyte solvent without salt (0 M). If a small stoichiometric surplus x of lithium is present in LTO, i.e., \(\text {Li}_{4+ x}\text {Ti}_5\text {O}_{12}\) with \(x>0,\) a thermodynamic chemical equilibrium would require \(\text {Li}^{+}\) to diffuse into the liquid phase. Since there are no negative ions available, no charge compensation is possible, causing a potential difference \(\Delta \phi\) between the two phases for an electrochemical equilibrium. Likewise, if electrolyte salt is present at high concentration but the solid phase does not contain any \(\text {Ti}^{3+}\) as formal negative charge, i.e., perfect stoichiometric LTO with \(x=0\) and without defects, no equilibration of the \(\text {Li}^{+}\) chemical potential is possible either. In between these border cases, a chemical equilibrium, which requires \(\Delta \phi =0,\) may be achievable. In fact, the electrolyte concentration-dependent \({}^7\text {Li}\) \(T_1\) data could be interpreted in the context of a transition between these two cases. Thereby, availability of \(\text {Ti}^{3+}\) in the solid and the structure of the LTO surface play a crucial role, and NMR may—indirectly—shed some light on the interplay between surface and bulk lithium in LTO.
In the absence of electrolyte salt, the electrolyte contains no ionic species, and charges near the LTO surface are only stabilized by the dielectric properties of the electrolyte solvent [40]. While it has been reported that polaronic charge in LTO has some energetic preference to be located near the surface [20], a more uniform distribution of polarons across the LTO volume would be entropically stabilized. The measured \({}^7\text {Li}\) \(T_1\) is representative for the balance between the two concurrent influences. \(T_1\) in the LTO bulk in the absence of an electrolyte salt is likely affected by some mobile polarons that ‘dress’ excess Li ions and, thereby, move as a formally neutral exotic polaronic atom [17]. Notice the similarity to a Rydberg atom, for which considerable diamagnetic shifts have been discussed [41], which may explain at least in part the observed difference of the spectra upfield and downfield of the electrolyte resonance [23, 30].
Once a small amount of electrolyte salt is added, \(\Delta \phi\) between LTO and electrolyte attracts negative charges from the electrolyte towards the LTO surface, causing a significant reduction of the Li-TFSI concentration in the electrolyte bulk, as detected by \({}^{19}\text {F}\) NMR of the supernatant. However, since no counter electrode is present, no macroscopic charge separation between cations and anions evolves. Charge separation occurs only microscopically within the surface complexion, while in the bulk electrolyte and in LTO, local charge neutrality is maintained. Assuming only a small amount of electrolyte salt, a chemical equilibrium would still require the extraction of Li ions into the electrolyte. A double layer forms with \(\text {TFSI}^{-}\) directly on the LTO surface to counteract the resulting potential difference. Charge compensation is achieved with \(\text {Li}^{+}\) ions from the electrolyte on the LTO averted side of the \(\text {TFSI}^{-}\) layer, leading to a surface dipole with the electronegative pole towards the LTO surface [42, 43]. This stabilizes \(\text {Li}^{+}\) dopant ions in LTO near the surface compared to the sample without electrolyte salt. A reduced \(\text {Li}^{+}\) dopant concentration with accompanying reduced paramagnetic polaron concentration in the LTO bulk is consistent with the observed increase of \({}^7\text {Li}\) \(T_1\) values in the low electrolyte concentration case, which suggests, according to the equilibrium in Eq. 1, a stabilization of polaronic charge in the surface complexion. Notice that this is not a static immobilization of polaronic charge on a particular Ti center, which would correspond to a physically implausible polaronic phase transition from a non-local to a localized behavior [44], but a gradual change of charge density delocalization.
As the Li-TFSI concentration is increased, \(\Delta \phi\) decreases until a chemical equilibrium between the two phases can be achieved, as shown schematically in Fig. 4. When the electrolyte concentration is increased further, maintaining a chemical equilibrium would require Li ions to diffuse into the LTO bulk. If defects are present in the surface complexion that can provide a charge for Li–polaron pairs, such a chemical equilibrium may be maintained over a certain electrolyte concentration range, and unpinned charges are locally compensated by \(\text {TFSI}^{-}.\) In this case, the \(\text {TFSI}^{-}\) surface layer is not stabilized by a potential difference anymore, but by the necessity for local charge compensation. The surface dipole vanishes, which is eased by the capability of \(\text {Li}^{+}\) to be located on the electrolyte as well as on the LTO side of the surface. Nonetheless, due to the absence of an external potential, a complex rearrangement of charge, as has been described for ionic liquids near surfaces [45], rather than a classical double layer may be the consequence.
If the electrolyte concentration is increased further, eventually \(\Delta \phi\) gets reestablished with inverted sign and a surface dipole with the electropositive pole towards the LTO surface is formed. In that case, \(\text {Li}^{+}\) ions are drawn to the LTO surface side of a \(\text {TFSI}^{-}\) layer. The surface layer is likely still primarily formed by \(\text {TFSI}^{-}\) ions, since \(\text {Li}^{+}\) can diffuse into LTO, yet remain in the complexion for charge neutrality. Electrostatically, this would stabilize defect \(\text {Li}^{+}\) in the bulk of LTO while, at the same time, stabilize polarons near the surface. The consequence could be a stabilization of neutral Li–polaron pairs near the surface, with polarons more localized on a particular \(\text {Li}^{+},\) or a minor charge separation between complexion and bulk. In both cases, surface pinning of polarons would be strengthened. Such an additional surface pinning of polarons would be consistent with a preferred polaron mobility along the LTO surface, as predicted theoretically [21]. Furthermore, it could be an explanation for the observed emergence of a two component \({}^7\text {Li}\) \(T_1\) distribution, with fast \(T_1\) in the shell and slow \(T_1\) in the core of LTO.
A model to explain the measured data is summarized in Fig. 5.

Equilibria between electrolyte, surface and bulk of LTO. a In the absence of an electrolyte salt, the electrolyte dimethylcarbonate has a minor effect on the surface–bulk equilibrium of lithium cations in LTO. b Lithium salt at a low concentration in the solution is adsorbed at the surface and shifts mobile polarons from the bulk to the surface. c Lithium cations from the electrolyte at higher concentration can diffuse with surface polarons into the bulk. Electrolyte anions are forced to interact with the surface
The surface complexion of pristine LTO contains some defects, which are charge compensated by polaronic \(\text {Ti}^{3+},\) to minimize the energy towards its environment. For stoichiometric LTO, numerical simulations have suggested that polarons show a weak energetic preference to be surface pinned [20]. Therefore their effect on NMR relaxation would be localized, and when observing bulk \({}^7\text {Li}\) \(T_1,\) only a minor influence is expected. However, in stoichiometric battery grade nano-LTO with a large surface area, surplus lithium cations may become more mobile if they move synchronously with polarons from the surface as a neutral entity [17]. In pristine LTO, there are fewer free lithium ions than polarons, because it can be assumed that oxygen vacancies in the solid are charge compensated by \(\text {Ti}^{3+}\) centers. These vacancies may interact with \(\text {Li}^{+}\) from the electrolyte.
\({}^7\text {Li}\) \(T_1\) showed a defined, continuous concentration dependent dispersion. As shown in Fig. 2c it was, however, not monotonic, suggesting that it is necessary to distinguish between a low concentration and a high concentration electrolyte regime. If a small concentration, here less than approximately 2 mmol/L, of lithium salt was added to the electrolyte, the anion concentration close the surface increased and formed additional negative charge carriers that stabilized mobile overstoichiometric \(\text {Li}^{+}\) in LTO near the surface. In this process, some spectral heterogeneity of \({}^7\text {Li}\) \(T_1\) was observed that may be interpreted by the formation of a core–shell structure. The core lost polaron concentration with increasing \(\text {TFSI}^{-}\) concentration in the electrolyte, which prevented paramagnetic relaxation processes and caused a minor increase of bulk \({}^7\text {Li}\) \(T_1\) in LTO. The polarons showed a stronger preference to be located in a shell on the LTO surface [20].
If a larger amount of lithium salt was added with a concentration above about 2 mmol/L, a critical concentration was reached that exerted an osmotic pressure on the solid, which is capable of taking up additional Li ions without significant structural adaptation. The \(\text {Li}^{+}\) now diffused from the electrolyte into the solid. Additional \(\text {Li}^{+}\) ions unpinned polarons from surface defects, forming electrically neutral, paramagnetic Li–polaron pairs with a reduced hopping barrier and, therefore, a higher diffusivity compared to a charged \(\text {Li}^{+}.\) This caused a homogeneous \({}^7\text {Li}\) \(T_1\) relaxation enhancement. The unpinned polarons from the surface were locally charge compensated by a \(\text {TFSI}^{-}.\)
At very high electrolyte concentrations, suitable defects in LTO are used up, and the formation of a less permeable core–shell configuration is indicated by \({}^7\text {Li}\) \(T_1\) data. Such a configuration may be stabilized by a space charge layer that forms a surface dipole, albeit direct evidence for such a mechanism is not apparent from the NMR data.
While the cases with low or very high electrolyte concentration require a better understanding of the surface structure and defects for an equilibrium model, a simplified model of the intermediate electrolyte concentration range may be attempted using the presented data. If we assume a chemical equilibrium between Li in the LTO bulk and in the electrolyte, then no electrical potential difference between the two phases exists, i.e., \(\Delta \phi = 0.\) \(\text {Li}^{+}\) in the complexion is in equilibrium as well, and it becomes possible to cascade the different equilibria such that eventually the Nernst potential between Li ions in the LTO bulk and in the electrolyte can be balanced by the redox potential of titanium ions. Let us consider the simplified equilibrium
where the subscripts (el), (cx), and (bulk) represent the electrolyte, the complexion, and the LTO bulk, respectively. Charge in each environment is balanced locally on the two sides of the equilibrium. For a qualitative discussion, an equilibrium constant based on concentrations rather than activities can be written as
Here, \([\text {Li}_{({\textrm{bulk}})}^{+}]\) is only the overstoichiometric lithium concentration. If an equilibrium is assumed between \(\text {Li}^{+}\) in the LTO bulk and in the electrolyte, as represented by the first term, \(K_{\textrm{Li}},\) then the second term, \(K_{\textrm{TFSI}},\) representing \(\text {TFSI}^{-}\) must form an equilibrium as well. Only \(K_{\textrm{Li}}\) is dependent on lithium concentrations, hence only this term is required for the discussion of \({}^7\text {Li}\) NMR data. It can further be simplified by remembering the assumption that in the LTO bulk, each \(\text {Ti}_{({\textrm{bulk}})}^{3+}\) is charge compensated by an overstoichiometric \(\text {Li}_{({\textrm{bulk}})}^{+},\) thus \([\text {Li}_{({\textrm{bulk}})}^{+}]=[\text {Ti}_{({\textrm{bulk}})}^{3+}].\) It is also assumed for the intermediate electrolyte concentration region that \([\text {Li}_{({\textrm{el}})}^+] \approx [\text {Li}_{({\textrm{el}})}^+]_0,\) where \([\text {Li}_{({\textrm{el}})}^+]_0\) is the concentration of the neat electrolyte. Finally, while both concentrations \([\text {Ti}_{({\textrm{cx}})}^{3+}]\) and \([\text {Ti}_{({\textrm{cx}})}^{4+}]\) in the complexion may change significantly, the amount of overstoichiometric Li diluted into the bulk is rather low, hence \([\text {Ti}_{({\textrm{bulk}})}^{4+}]\) can be approximated by the stoichiometric concentration \([\text {Ti}_{({\textrm{bulk}})}^{4+}]_0\) and is, therefore, constant. The simplified equilibrium to be considered then becomes
If constant terms are combined into \(\tilde{K} = \sqrt{K [\text {Ti}_{({\textrm{bulk}})}^{4+}]_0 / K_{\textrm{TFSI}}} ,\) then the bulk polaron concentration becomes
The link with the experimentally observable \(T_1\) relaxation time constants can be made by assuming the presence of mobile paramagnetic polarons in the LTO bulk with concentration [\(\text {Ti}_{({\textrm{bulk}})}^{3+}\)] and modeling the observed relaxation rate \(R_1=1/T_1\) as
where \(R_{1,0}\) is the relaxation rate in the absence of paramagnetic species, and \(\rho\) is an empirical paramagnetic relaxivity parameter that is characteristic for a particular material, temperature, and magnetic field strength. Results are shown in Fig. 6.

Fit of \({}^7\text {Li}\) \(T_1\) relaxation time distribution maximum in LTO vs. electrolyte concentration using Eq. 5 for the downfield (Green) and the upfield (Orange) spectral regions. For both data sets, four data points were used for fitting (diamonds). The data points shown as circles were not used for the fit. Fitted parameters were \(\rho \tilde{K} = 2.14 \pm 0.06 \times 10^{-3}\,{{\text {s}^{-1}}\,(\text {mM})^{-0.5}}\) and \(R_{1,0} = 7.8 \pm 0.5 \times 10^{-3}\,{{\text {s}^{-1}}}\) for the downfield, and \(\rho \tilde{K} = 1.81 \pm 0.02 \times 10^{-3}\,{{\text {s}^{-1}}\, (\text {mM})^{-0.5}}\) and \(R_{1,0} = 9.9 \pm 0.3 \times 10^{-3}\,{{\text {s}^{-1}}}\) for the upfield spectral region. For both fits, the coefficient of determination was above 0.999 (color figure online)
The model is capable of fitting the intermediate concentration region well, indicating that the model assumptions are sufficiently justified. The low concentration region, however, cannot be represented adequately, which is in line with the suggestion of a qualitatively different equilibrium. Fitting is possible in a robust manner, with \(T_{1,0} = 1/R_{1,0}\) on the same order as reported for sodium containing electrolyte, where the cation is not able to enter the LTO matrix [14].
Despite the apparently homogeneous \(T_1\) time constants at intermediate electrolyte concentration, upfield and downfield regions of the spectrum provided different fitting parameters. Using high resolution magic angle spinning (MAS) \({}^7\text {Li}\) NMR of overlithiated LTO, an upfield Knight shift or paramagnetic shift was observed due to the formation of paramagnetic \(\text {Ti}^{3+}\) [23, 30]. While MAS NMR experiments would be necessary to confirm such a correlation, the observed variations of the fitted parameters, with a shorter \(T_{1,0}\) for the upfield region, is consistent with such an origin.
5 Conclusions
The \({}^7\text {Li}\) \(T_1\) relaxation behavior of LTO in the presence of lithium-salt-containing aprotic electrolytes was reported as a function of electrolyte concentration. It was shown that there is a defined relationship between the bulk \({}^7\text {Li}\) \(T_1\) relaxation time in LTO and the lithium ion concentration in the surrounding electrolyte. At electrolyte concentrations larger than about 2 mmol/L, the relaxation time in the solid state shortened with increasing lithium ion concentration in the electrolyte. This could be explained by the lithium interdiffusion equilibrium that occurs between the LTO solid and the electrolyte. Lithium ions diffuse into the solid, which is possible because polaronic electrons can be released from existing defects in the LTO complexion. The Li ions and the polarons move as a neutral quasiparticle, allowing local charge balance for the newly introduced \(\text {Li}^{+}.\) The positive defects at the LTO surface are charge compensated by anions from the electrolyte, which are not able to enter the solid. If the concentration of the lithium salt is below 2 mmol/L, lithium ions no longer diffuse into the bulk. Instead, anions are adsorbed at the LTO surface, where they form a space charge layer that acts, together with the \(\text {Li}^{+}\) ions from the electrolyte, as a surface dipole with its electronegative pole pointing towards the LTO to stabilize the defects. A model based on a core–shell structure was presented. This leads to an \({}^7\text {Li}\) NMR frequency dependent relaxation behavior at low electrolyte concentration, with faster relaxation in the upfield region and slower relaxation in the downfield region, while spectrally more homogeneous relaxation was observed at concentrations larger than 10 mmol/L and up to about 250 mmol/L.
An empirical equilibrium model was devised for the intermediate electrolyte concentration region, which showed adequate fits of the experimental data. It is consistent with and further refines previously established connections with pseudocapacitance, implying an adsorption pseudocapacitance at low and possibly very high concentrations, and redox or intercalation pseudocapacitance in intermediate concentration regions [46, 47]. Therefore, it correlates with defects in the material [48].
A tool for investigating the influence of doping, either deliberate or accidental, on properties of battery materials was presented, using the example of battery grade LTO. The presented approach may prove useful for material development, either empirically as presented here, or based on a more physics-based analysis in combination with additional experiments and numerical simulations. In particular, mutual stimulation may occur by combining such measurements with simulations. To extract more physically meaningful parameters from measurements, certain constraints are required. Recent methodological developments in atomistic simulations of LTO and its surface suggest that such constraints may be obtained numerically [19, 49]. At the same time, the presented experiments may become a realistic target for direct simulations in the future. Furthermore, microstructure-resolved simulations, which strongly depend on the availability of a large number of accurate physical parameters [50], may benefit from measurements of battery-relevant material combinations that avoid the complexity of an electrochemical cell. However, substantial hurdles still exist. As one example in the case of LTO or, more generally, battery electrode materials, the relaxivity parameter \(\rho\) may be sensitively dependent on the \(\text {Li}^{+}\) concentration in LTO, since the Li-ion mobility in the material changes as a function of overlithiation [31]. A more detailed analysis may be possible and worthwhile, but would require a more involved model of relaxation that takes into account both the Li ion mobility dependence on the degree of overlithiation and the additional relaxation effects by the paramagnetic polaronic charges. For both of these problems, no quantitative theoretical basis exist. For its development, various magnetic resonance techniques need to be combined, including MAS NMR, EPR, or field-cycling relaxometry, and potentially supplemented by electrochemical tests [51]. If successful, a more direct interpretation of presented results and their subsequent use for material optimization may be envisioned.
Data Availability
The data sets used and analyzed during the current study are available from the corresponding author on reasonable request.
References
M. Weiss, R. Ruess, J. Kasnatscheew, Y. Levartovsky, N.R. Levy, P. Minnmann, L. Stolz, T. Waldmann, M. Wohlfahrt-Mehrens, D. Aurbach, M. Winter, Y. Ein-Eli, J. Janek, Fast charging of lithium-ion batteries: a review of materials aspects. Adv. Energy Mater. 11, 2101126 (2021). https://doi.org/10.1002/aenm.202101126
P. Simon, Y. Gogotsi, B. Dunn, Where do batteries end and supercapacitors begin? Science 343, 1210–1211 (2014). https://doi.org/10.1126/science.1249625
Y. Gogotsi, R.M. Penner, Energy storage in nanomaterials—capacitive pseudocapacitive, or battery-like? ACS Nano 12, 2081–2083 (2018). https://doi.org/10.1021/acsnano.8b01914
M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P.-L. Taberna, C.P. Grey, B. Dunn, P. Simon, Efficient storage mechanisms for building better supercapacitors. Nat. Energy 1, 16070 (2016). https://doi.org/10.1038/NENERGY.2016.70
M.R. Lukatskaya, B. Dunn, Y. Gogotsi, Multidimensional materials and device architectures for future hybrid energy storage. Nat. Commun. 7, 12647 (2016). https://doi.org/10.1038/ncomms12647
D.P. Dubal, O. Ayyad, V. Ruiz, P. Gomez-Romero, Hybrid energy storage: the merging of battery and supercapacitor chemistries. Chem. Soc. Rev. 44, 1777–1790 (2015). https://doi.org/10.1039/c4cs00266k
G.G. Amatucci, F. Badway, A. Du Pasquier, T. Zheng, An asymmetric hybrid nonaqueous energy storage cell. J. Electrochem. Soc. 148(8), 930–939 (2001). https://doi.org/10.1149/1.1383553
J.M. Feckl, K. Fominykh, M. Döblinger, D. Fattakhova-Rohlfing, T. Bein, Nanoscale porous framework of lithium titanate for ultrafast lithium insertion. Angew. Chem. Int. Ed. 51, 7459–7463 (2012). https://doi.org/10.1002/anie.201201463
C. Franchini, M. Reticcioli, M. Setvin, U. Diebold, Polarons in materials. Nat. Rev. Mater. 6, 560–586 (2021). https://doi.org/10.1038/s41578-021-00289-w
P. Jakes, J. Granwehr, H. Kungl, R.-A. Eichel, Mixed ionic electronic conducting \(\text{ Li}_4\text{ Ti}_5\text{ O}_{12}\) as anode material for lithium ion batteries with enhanced rate capability—impact of oxygen non-stoichiometry and aliovalent \(\text{ Mg}^{2+}\)-doping studied by electron paramagnetic resonance. Z. Phys. Chem. 229, 1439–1450 (2015). https://doi.org/10.1515/zpch-2015-0618
J.M. Griffin, A.C. Forse, C.P. Grey, Solid-state NMR studies of supercapacitors. Solid State Nucl. Magn. Reson. 74–75, 16–35 (2016). https://doi.org/10.1016/j.ssnmr.2016.03.003
B. Blümich, Introduction to compact NMR: a review of methods. TRAC Trend. Anal. Chem. 83, 2–11 (2016). https://doi.org/10.1016/j.trac.2015.12.012
S. Anferova, V. Anferov, J. Arnold, E. Talnishnikh, M.A. Voda, K. Kupferschlaeger, P. Bluemler, C. Clauser, B. Bluemich, Improved Halbach sensor for NMR scanning of drill cores. Magn. Reson. Imaging 25, 474–480 (2007). https://doi.org/10.1016/j.mri.2006.11.016
P.P.M. Schleker, C. Grosu, M. Paulus, P. Jakes, R. Schlögl, R.-A. Eichel, C. Scheurer, J. Granwehr, Electrolyte contact changes nano-\(\text{ Li}_4\text{ Ti}_5\text{ O}_{12}\) bulk properties via surface polarons. Commun. Chem. 6, 113 (2023). https://doi.org/10.1038/s42004-023-00913-6
M. Mohammadi, S. Benders, A. Jerschow, Nuclear magnetic resonance spin-lattice relaxation of lithium ions in aqueous solution by NMR and molecular dynamics. J. Chem. Phys. 153, 184502 (2020). https://doi.org/10.1063/5.0026450
H. Oberhofer, K. Reuter, J. Blumberger, Charge transport in molecular materials: an assessment of computational methods. Chem. Rev. 117, 10319–10357 (2017). https://doi.org/10.1021/acs.chemrev.7b00086
H.D. Luong, T.L. Tran, V.B.T. Phung, V.A. Dinh, Small polaron transport in cathode materials of rechargeable ion batteries. J. Sci. Adv. Mater. Devices 7(1), 100410 (2022). https://doi.org/10.1016/j.jsamd.2021.100410
T. Maxisch, F. Zhou, G. Ceder, Ab initio study of the migration of small polarons in olivine \(\text{ Li}_{x}\text{ FePO}_4\) and their association with lithium ions and vacancies. Phys. Rev. B (2006). https://doi.org/10.1103/PhysRevB.73.104301
M. Kick, C. Grosu, M. Schuderer, C. Scheurer, H. Oberhofer, Mobile small polarons qualitatively explain conductivity in lithium titanium oxide battery electrodes. J. Phys. Chem. Lett. 11, 2535–2540 (2020). https://doi.org/10.1021/acs.jpclett.0c00568
M. Kick, C. Scheurer, H. Oberhofer, Formation and stability of small polarons at the lithium-terminated \(\text{ Li}_4\text{ Ti}_5\text{ O}_{12}\) (LTO) (111) surface. J. Chem. Phys. 153, 144701 (2020). https://doi.org/10.1063/5.0021443
M. Kick, C. Scheurer, H. Oberhofer, Polaron-assisted charge transport in Li-ion battery anode materials. ACS Appl. Energy Mater. 4, 8583–8591 (2021). https://doi.org/10.1021/acsaem.1c01767
G.C. Borgia, R.J.S. Brown, P. Fantazzini, Uniform-penalty inversion of multiexponential decay data. J. Magn. Reson. 132, 65–77 (1998). https://doi.org/10.1006/jmre.1998.1387
M.F. Graf, H. Tempel, S.S. Köcher, R. Schierholz, C. Scheurer, H. Kungl, R.-A. Eichel, J. Granwehr, Observing different modes of mobility in lithium titanate spinel by nuclear magnetic resonance. RSC Adv. 7, 25276–25284 (2017). https://doi.org/10.1039/c7ra01622k
J. Granwehr, P.J. Roberts, Inverse Laplace transform of multidimensional relaxation data without non-negativity constraint. J. Chem. Theory Comput. 8, 3473–3482 (2012). https://doi.org/10.1021/ct3001393
J.W. Eaton, D. Bateman, S. Hauberg, R. Wehbring, GNU Octave version 4.0.0 manual: a high-level interactive language for numerical computations (2015). http://www.gnu.org/software/octave/doc/interpreter. Accessed 5 Sept 2023
S. Merz, P. Jakes, S. Taranenko, R.-A. Eichel, J. Granwehr, Dynamics of \([\text{ Pyr}_{13}\)][\(\text{ Tf}_2\text{ N}]\) ionic liquid confined to carbon black. Phys. Chem. Chem. Phys. 21, 17018–17028 (2019). https://doi.org/10.1039/c9cp02651g
M.C. Paulus, A. Paulus, P.P.M. Schleker, P. Jakes, R.-A. Eichel, P. Heitjans, J. Granwehr, Experimental evidence for the relaxation coupling of all longitudinal \({}^7\text{ Li }\) magnetization orders in the superionic conductor \(\text{ Li}_{10}\text{ GeP}_{2}\text{ S}_{12}\). J. Magn. Reson. 303, 57–66 (2019). https://doi.org/10.1016/j.jmr.2019.04.006
M.C. Paulus, M.F. Graf, P.P.R.M.L. Harks, A. Paulus, P.P.M. Schleker, P.H.L. Notten, R.-A. Eichel, J. Granwehr, Investigation of the Li-ion conduction behavior in the \(\text{ Li}_{10}\text{ GeP}_{2}\text{ S}_{12}\) solid electrolyte by two-dimensional \(t_1\)-spin alignment echo correlation NMR. J. Magn. Reson. 294, 133–142 (2018). https://doi.org/10.1016/j.jmr.2018.07.008
M. Vijayakumar, S. Kerisit, K.M. Rosso, S.D. Burton, J.A. Sears, Z. Yang, G.L. Graff, J. Liu, J. Hu, Lithium diffusion in \(\text{ Li}_4\text{ Ti}_5\text{ O}_{12}\) at high temperatures. J. Power Sources 196, 2211–2220 (2011). https://doi.org/10.1016/j.jpowsour.2010.09.060
M. Wagemaker, D.R. Simon, E.M. Kelder, J. Schoonman, C. Ringpfeil, U. Haake, D. Lützenkirchen-Hecht, R. Frahm, F.M. Mulder, A kinetic two-phase and equilibrium solid solution in spinel \(\text{ Li}_4+x\text{ Ti}_5\text{ O}_12\). Adv. Mater. 18, 3169 (2006). https://doi.org/10.1002/adma.200601636
W. Schmidt, P. Bottke, M. Sternad, P. Gollob, V. Hennige, M. Wilkening, Small change-great effect: steep increase of li ion dynamics in \(\text{ Li}_4\text{ Ti}_5\text{ O}_{12}\) at the early stages of chemical li insertion. Chem. Mater. 27, 1740–1750 (2015). https://doi.org/10.1021/cm504564k
S. Rodts, D. Bytchenkoff, Structural properties of 2D NMR relaxation spectra of diffusive systems. J. Magn. Reson. 205, 315–318 (2010). https://doi.org/10.1016/j.jmr.2010.04.021
D. Columbus, V. Arunachalam, F. Glang, L. Avram, S. Haber, A. Zohar, M. Zaiss, M. Leskes, Direct detection of lithium exchange across the solid electrolyte interphase by \({}^7\text{ Li }\) chemical exchange saturation transfer. J. Am. Chem. Soc. 144, 9836–9844 (2022). https://doi.org/10.1021/jacs.2c02494
J.M. Griffin, A.C. Forse, H. Wang, N.M. Trease, P.-L. Taberna, P. Simon, C.P. Grey, Ion counting in supercapacitor electrodes using NMR spectroscopy. Faraday Discuss. 176, 49–68 (2014). https://doi.org/10.1039/C4FD00138A
F. Criscuolo, M.I.N. Hanitra, I. Taurino, S. Carrara, G. De Micheli, All-solid-state ion-selective electrodes: a tutorial for correct practice. IEEE Sens. J. 21, 22143–22154 (2021). https://doi.org/10.1109/JSEN.2021.3099209
P.R. Cantwell, M. Tang, S.J. Dillon, J. Luo, G.S. Rohrer, M.P. Harmer, Grain boundary complexions. Acta Mater. 62, 1–48 (2014). https://doi.org/10.1016/j.actamat.2013.07.037
Z. Chen, I. Belharouak, Y.-K. Sun, K. Amine, Titanium-based anode materials for safe lithium-ion batteries. Adv. Funct. Mater. 23, 959–969 (2013). https://doi.org/10.1002/adfm.201200698
A. Grimaud, W.T. Hong, Y. Shao-Horn, J.-M. Tarascon, Anionic redox processes for electrochemical devices. Nat. Mater. 15, 121–126 (2016). https://doi.org/10.1038/nmat4551
T. Luo, S. Abdu, M. Wessling, Selectivity of ion exchange membranes: a review. J. Membr. Sci. 555, 429–454 (2018). https://doi.org/10.1016/j.memsci.2018.03.051
J.P. Souza, A.A. Kornyshev, M.Z. Bazant, Polar liquids at charged interfaces: a dipolar shell theory. J. Chem. Phys. 156, 244705 (2022). https://doi.org/10.1063/5.0096439
V. Ovsiannikov, S. Goossev, Diamagnetic shift and splitting of Rydberg levels in atoms. Phys. Scr. 57, 506–513 (1998). https://doi.org/10.1088/0031-8949/57/4/007
D. Cahen, A. Kahn, Electron energetics at surfaces and interfaces: concepts and experiments. Adv. Mater. 15, 271–277 (2003). https://doi.org/10.1002/adma.200390065
A.A. Kornyshev, R. Qiao, Three-dimensional double layers. J. Phys. Chem. C 118, 18285–18290 (2014). https://doi.org/10.1021/jp5047062
B. Gerlach, H. Löwen, Analytical properties of polaron systems or: do polaronic phase transitions exist or not? Rev. Mod. Phys. 63, 63–90 (1991). https://doi.org/10.1103/RevModPhys.63.63
M. McEldrew, Z.A.H. Goodwin, A.A. Kornyshev, M.Z. Bazant, Theory of the double layer in water-in-salt electrolytes. J. Phys. Chem. Lett. 9, 5840–5846 (2018). https://doi.org/10.1021/acs.jpclett.8b02543
S. Fleischmann, J.B. Mitchell, R. Wang, C. Zhan, D.-E. Jiang, V. Presser, V. Augustyn, Pseudocapacitance: from fundamental understanding to high power energy storage materials. Chem. Rev. 120, 6738–6782 (2020). https://doi.org/10.1021/acs.chemrev.0c00170
V. Augustyn, P. Simon, B. Dunn, Pseudocapacitive oxide materials for high-rate electrochemical energy storage. Energy Environ. Sci. 7, 1597–1614 (2014). https://doi.org/10.1039/C3EE44164D
H.-S. Kim, J.B. Cook, H. Lin, J.S. Ko, S.H. Tolbert, V. Ozolins, B. Dunn, Oxygen vacancies enhance pseudocapacitive charge storage properties of \(\text{ MoO}_{3-\text{ x }}\). Nat. Mater. 16, 454–460 (2017). https://doi.org/10.1038/NMAT4810
S.S. Köcher, P.P.M. Schleker, M.F. Graf, R.-A. Eichel, K. Reuter, J. Granwehr, C. Scheurer, Chemical shift reference scale for Li solid state NMR derived by first-principles DFT calculations. J. Magn. Reson. 297, 33–41 (2018). https://doi.org/10.1016/j.jmr.2018.10.003
V. De Lauri, L. Krumbein, S. Hein, B. Prifling, V. Schmidt, T. Danner, A. Latz, Beneficial effects of three-dimensional structured electrodes for the fast charging of lithium-ion batteries. ACS Appl. Energy Mater. 4, 13847–13859 (2021). https://doi.org/10.1021/acsaem.1c02621
M. Schleutker, J. Bahner, C.-L. Tsai, D. Stolten, C. Korte, On the interfacial charge transfer between solid and liquid \(\text{ Li}^{+}\) electrolytes. Phys. Chem. Chem. Phys. 19, 26596–26605 (2017). https://doi.org/10.1039/c7cp05213h
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was partially funded by the German Federal Ministry of Education and Research (BMBF) within the project AdamBatt (FKZ 13XP0305A).
Author information
Authors and Affiliations
Contributions
PPMS designed and conducted the experiments. PPMS and JG performed the data analysis. PPMS and JG wrote the original draft with input from RAE, and all authors reviewed the manuscript. RAE acquired funding.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Dedicated to Bernhard Blümich on the occasion of his 70th birthday.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schleker, P.P.M., Eichel, RA. & Granwehr, J. Revealing the Equilibrium of Lithium Cations Across a Solid–Electrolyte Interface by \(T_1\) NMR Relaxation. Appl Magn Reson 54, 1463–1480 (2023). https://doi.org/10.1007/s00723-023-01609-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-023-01609-9