
Abstract
The impact of alcohol on the body can be investigated with NMR spectroscopy in vitro, which can detect a wide range of metabolites but preparing samples includes tissue biopsy. Blood sampling is less invasive, but blood metabolic content might not reflect the changes occurring in other tissues. Thus, this study aimed to investigate the liver, brain, and serum metabolism and evaluate the link between tissues and serum metabolic content. Two experimental groups with ten outbred rats each were provided intragastrically with water (control group) and 50% ethanol solution (alcohol group) for 28 days. 1H NMR spectroscopy in vitro was performed on the brain cortex, liver, and serum samples. Student’s t test with Holm–Bonferroni correction was used to investigate significant differences between groups. Partial least-squares discriminant analysis (PLS-DA) and two-way ANOVA were performed to compare liver and serum, brain and serum. In all, 38, 37, and 21 metabolites were identified in the liver, brain, and serum samples, respectively. Significant differences for three metabolites were found in the liver (alanine, proline, and glutathione, p < 0.002) and four in serum (lactate, betaine, acetate, and formic acid, p < 0.002) were detected between the control and alcohol groups. The contents of glucose, betaine, and isoleucine were correlated (r > 0.65) between serum and liver samples. PLS-DA determined separation between all tissues (p < 0.001) and between control and alcohol groups only for liver and serum (p < 0.001). Alcohol had a more substantial effect on liver and serum metabolism than on the brain.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
An organism develops a severe response to acute alcohol intoxication. In contrast, chronic intoxication develops imperceptibly in the early stages of the disease. However, regular alcohol consumption is more common. It may affect metabolic processes and induce functional defects in individual organs and systemic changes in the whole body.
As the primary organ for detoxication, the liver bears the main toxic load [1]. The harmful impact of alcohol on liver metabolism has been reported in numerous publications [2, 3]. A limited amount of toxic compounds can be neutralized in the liver within a unit of time; thus, poisonous alcohol metabolism products are freely transferred by the bloodstream to other organs. Therefore, the brain and nervous system are the victims of alcohol too. The alcohol-related cognitive disorders include partial and complete memory blackouts [4], peripheral neuropathy [5], dementia [6]. Besides, alcohol promotes the neurons’ death and inhibits the proliferation of new cells [7].
Fewer studies have reported alcohol’s effect on brain or serum metabolism [8, 9] than having described the impact of alcohol on the liver [10,11,12,13]. The changes in blood composition are particularly interesting since the blood sampling procedure is minimally invasive. Therefore, blood tests can be performed regularly to diagnose or monitor disease progression or treatment efficacy. New methods and biomarkers are needed to identify alcoholic liver diseases, as currently, the diagnosis is often confirmed by an invasive liver biopsy [14]. Thus, this study aims first to investigate the metabolism of liver, brain, and serum and, secondly, to evaluate the link between tissues and serum metabolic content using 1H nuclear magnetic resonance (NMR) spectroscopy in vitro.
Metabolism can be studied using two main methods: NMR spectroscopy and mass spectrometry. In the current study, a metabolomic approach based on 1H NMR spectroscopy in vitro was used because the results of the NMR spectroscopy in vitro studies can be translated or compared to in vivo magnetic resonance spectroscopy studies.
In our earlier work [15], we evaluated the impact of a low alcohol dose (2–3 g/kg) only on rat’s liver. The alcohol consumption stimulated the proliferation of hepatocytes and changed the concentrations in the range of metabolites (e.g., leucine, valine, choline, etc.). The presented study aims to extend the previous result by including the brain and serum as alcohol targets.
Previously, the simultaneous effects of alcohol on the liver, brain, and blood of rats were investigated solely by NMR spectroscopy [9]. Furthermore, both acute and chronic alcohol exposure were considered (a single alcohol dose, 5 g ethanol/kg body weight, and a 4-day binge ethanol protocol, 8.6 g/kg per day). The metabolic profile of the brain did not show any statistically significant metabolic changes between experimental groups. A significant decrease in the concentration of glucose, alanine, and lactate in the liver and serum metabolic profiles for both acute and chronic alcohol exposure was reported. In addition, Obernier et al. reported similar findings, except for the changes in glucose concentrations [16].
In comparison with the work of Nicholas et al. [9] we added additional conditions for alcohol intake to this study; in particular, the duration of chronic alcohol consumption (8–10 g/kg per day) was increased from 4 to 28 days. A comparative analysis was performed to find an association between tissues and serum metabolome under alcohol exposure.
2 Materials and Methods
2.1 Experimental Animals and Manipulation
Twenty outbred Sprague–Dawley rats were provided by the Center for Genetic Resources of Laboratory Animals at the Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences (RFMEFI62119X0023). The rats were 12–13 weeks old at the beginning of the experiment. During the investigation, animals were housed in pairs in individually ventilated cages with an artificial day-night regimen (OptiRAT cage; Charles River Laboratories, Chatillon-Sur-Chalaronne, France; 14 h light–10 h darkness, temperature 22–24 °C, and humidity 40–50%). The food and water supply were provided ad libitum (Chara SPF granulated forage for laboratory rodents).
Chronic alcohol liver pathology was modeled for 4 weeks. The animals were divided into two experimental groups, with ten animals in each:
-
1.
Control group C: animals received intragastrically 5 ml of water twice a day (9:00 a.m. and 9:00 p.m.).
-
2.
Alcohol group A: animals received intragastrically 5 ml of alcohol twice a day (9:00 a.m. and 9:00 p.m.).
Water and alcohol were administered intragastrically using a rat intragastric probe. The daily alcohol dose was gradually increased from 40% at the first day, 45% at the second day, and 50% ethanol solution was used after that. This gradual increase in alcohol intake was used to avoid stomach inflammation.
After 4 weeks, rats were sacrificed by decapitation. The right liver lobe and brain cortex from each animal were collected. Mixed arteriovenous blood was taken and centrifuged (3000 rpm, 15 min). The resulting supernatant and the tissues were frozen at − 80 °C and stored there until sample preparation (for 1 month, Kelvinator, SANYOMDF-594, SANYO Electric Co., Japan).
2.2 Preparation of Liver, Brain, and Serum Samples for 1 H NMR Spectroscopy In Vitro
Tissue samples (liver and brain) were removed from the freezer the day before the NMR examination. A portion of the tissue was cut and homogenized using the ethanol extraction procedure (70 mg of tissue, 700 μl of 80% ethanol solution). Hydrophobic and hydrophilic metabolites were separated using a chloroform-water mixture (1:1 by volume) followed by dehydration of the hydrophilic part over 12 h (CentriVap Solvent Concentrator Systems, Labconco, USA). The detailed protocol was published previously [15], and the necessity of the steps to be performed was also described earlier [17, 18].
Serum samples were removed from the freezer and thawed at room temperature (21 °C, 5 min). Then, 150 μl of serum was carefully mixed with 700 μl of 80% ethanol. The remaining steps were identical to the tissue preparation protocol described above.
The ethanol extraction for the liver, brain, and serum was performed on ice (4 °C).
For the NMR spectroscopy examination, the water-soluble metabolites were dissolved in D2O-based phosphate-buffered saline (99.9%, Sigma-Aldrich, 600 μl). The resulting solution was mixed (5 min, BioSan Vortex V-1, BioSan, Latvia) and sonicated until the metabolites had wholly dissolved (20 min, Q700, Qsonica, USA).
2.3 1H NMR Spectroscopy
All 1H NMR spectra were obtained at 298 K using a high-resolution 700 MHz NMR spectrometer (Avance III HD, Bruker, Billerica, Massachusetts, US). A single-pulse sequence with a 7.7-μs-long, rectangular, nonselective radiofrequency 900 pulse was used for signal acquisition. Low-power continuous-wave irradiation was applied during the prescan delay for water suppression. The free induction decay was collected using four dummy scans and 200 accumulation scans; the delay time between the scans was 17 s. One full measurement took 68 min in total.
2.4 Processing of 1 H NMR Spectra
The interpretation of 1H NMR spectra was based on the literature analysis [9, 19, 20]. The metabolites derived from the literature were cross-checked with the NMR spectra obtained using metabolomic databases. The most intensive 1H NMR peaks were matched to the corresponding metabolites based on the spectra of individual metabolites (HMDB http://www.hmbd.ca; BMRDB http://www.bmrb.wisc.edu).
The multipoint baseline correction was applied to all spectra before quantifying the metabolites (MestReNova v9.0, Mestrelab Research, Santiago de Compostela, Spain). The identified multiplets were manually integrated. The resulting integrated intensity of each multiplet or part of a multiplet without overlapping with other signals was used as a measure of metabolic concentration. If more than one of the nonoverlapping multiplets corresponded to the same metabolite, then the sum of the integrated intensities was evaluated. The resulting metabolic concentrations were normalized by the sum of all identified metabolites in each tissue and serum sample. The normalization process was performed to eliminate any differences in initial tissue weight and inhomogeneity.
2.5 Statistical Processing
The following statistical analysis was performed for all assigned metabolites (Statistica 6.0, TIBCO Software Inc., Palo Alto, USA): 1. The Kolmogorov–Smirnov test was performed to confirm the normal distribution of metabolomic concentrations among the species. 2. Student’s t test was used to reveal differences between means of metabolic concentrations related to group A and C. 3. The Holm–Bonferroni correction was applied to a significance level of Student’s t test (p = 0.05) to avoid false-positive significant differences between metabolites from group A and C. 4. The Pearson correlation coefficient (r) was calculated to find the relationship between serum and liver metabolism, and serum and brain metabolism. 5. The number of initial variables (metabolites) was reduced with partial least-squares discriminant analysis (PLS-DA), which allows maximizing the separation between experimental groups [21]. The PLS-DA builds a linear regression model by projecting the initial variables (metabolites) to a new space, with Y as a categorical variable [22]. PLS-DA was performed separately for the group A and C of liver and serum and both groups of brain and serum. Then two-way ANOVA (on tissue and experimental group) was performed, with Y values as the dependent variable, to assess the distribution of experimental groups in the space of Y axes.
Metabolic concentrations are represented as a mean value over the experimental groups ± standard deviation.
3 Results
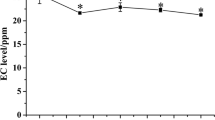
First of all, during the first week of the experiment, the weight of animals in group A was decreased from 467 ± 22.7 g to 426.9 ± 43.18 (p < 0.001, Fig. 1). However, then animals increased their weight, and by the end of the experiment, they had gained their weight to 484.4 g. The constant increase of the animal weight in the control group was observed from 433.3 g at week 1 to 481.4 g at week 4 (p value < < 0.01). The animal weight before the metabolomic analysis (week 4) did not differ statistically between experimental groups (group A = 484.8 g, group C = 481.4 g, p value = 0.84).

Average animal weight in alcohol and control groups measured at the end of the experimental week. The whiskers indicate the standard deviation within the group. The * symbol indicates the statistically significant differences at p < 0.01
In the 1H NMR spectra collected, the broadening of the multiplets often caused by macromolecules was not observed (proteins, lipids, nucleic acids, etc., were removed from the sample during the sample preparation). The representative 1H NMR spectra of animals from group C are shown in Fig. 2 and from group A in Figure S1 (Supplementary Information). In all, 38 liver, 37 brain, and 21 serum metabolites were identified (Table S1, Supplementary Information).

Representative 1H NMR spectra (0.9–3.9 ppm) of the liver, brain, and serum extracts derived from the control group. 1—leucine; 2—valine; 3—isoleucine; 4—3-hydroxybutyric acid; 5—aspartate; 6—choline; 7—phosphocholine; 8—betaine, 9—proline
Student’s t test was applied to the individual metabolite concentrations obtained. Taking into account the Holm–Bonferroni correction for 38 liver, 37 brain and 21 serum metabolites to the significance level, only three metabolites in the liver: alanine (p = 0.0013), proline (p < 0.001), and glutathione (p = 0.0011) and four in serum: lactate (p < 0.001), betaine (p < 0.001), acetate (p < 0.001), and formic acid (p = 0.0011) were found to be different between experimental groups (Table 1, Fig. 3). Besides, methionine was found to be diminished for the alcohol group in the range of reliability (p value = 0.019).

The graphic representation shows the metabolic changes between control and alcohol groups found in liver and serum extracts. The line represents a median, cross—a mean, and box is a first and third quartile. Whiskers are the minimum and maximum values in each experimental group
Fourteen metabolites (Table 1) were analyzed in both serum and liver, only three were correlated between tissues: alpha-d-glucose (r = 0.74), betaine (r = 0.73) and isoleucine (r = 0.65). Fifteen metabolites were analyzed in both serum and brain samples (Table 1). However, none of them was significantly correlated (r < 0.36) between tissues.
As a result of the PLS-DA analysis, two categorical variables Y1 and Y2, were obtained. Y1 and Y2 represent linear combinations of common metabolites between considered tissues. Each animal has their own coordinate at the space of Y variables based on its metabolic content. These coordinates can be presented by the 2D plot (Fig. 4). Figure 4, left, represents the individual metabolic contribution to the PLS-DA space; each point corresponds to one animal. The right part represents the mean values of these metabolic contributions averaged over the experimental group. Loading weights of the levels of metabolites on PLS-DA model “Liver vs. Serum” and “Brain vs. Serum” were R2Y = 41% and 0.68%, respectively (R2Y reflects the fraction of the variation of the Y variables explained by the model).

Distribution of experimental animal groups in the space of the Y1 and Y2 axes obtained by analyzing metabolites in liver and serum samples (a) and brain and serum samples (b) by partial least-squares discriminant analysis. Note that the liver and serum samples are separated in the axes Y2 by alcohol intake and brain samples are not
In PLS-DA model “Liver vs. Serum”, two-way ANOVA analysis showed that tissue type has a significant influence on the Y1 axis (F1,14 = 504.75, p value < < 0.01), but alcohol consumption does not (F1,14 = 0.16, p value = 0.69). In contrast to the Y1, Y2 was influenced by an alcohol consumption (F1,14 = 25.78, p value < < 0.01), but not by the tissue type (F1,14 = 0.01, p value = 0.93).
In PLS-DA model “Brain vs. Serum “, two-way ANOVA analysis showed again that tissue type has significant impact on Y1 axis (F1,15 = 2073.63, p value < < 0.01), but alcohol consumption does not (F1,15 = 0.001, p value = 0.98). Opposite results were observed for the Y2 axis, the alcohol consumption has a significant impact (F1,15 = 45.11, p value < < 0.01), and the tissue type does not (F1,15 = 0.00001, p value = 0.997).
According to the group separation in the Y1 and Y2 space and results of two-way ANOVA, we can associate the Y1 with a tissue type and Y2 with alcohol consumption.
4 Discussion
Overall, the impact of alcohol on rat metabolism found here was relatively weak. Only a few metabolic concentrations were different between the control and alcohol groups in the liver and serum (Fig. 3), with no changes seen in the brain. Adaptation to the toxic load may have taken place during the second and third weeks of the experiment, as animal weight in the alcohol group decreased first, then increased and became statistically not different from the weight of animals in the control group. However, other metabolic pathways, which persisted to the fourth week of alcohol exposure, might have been affected by the alcohol.
4.1 Liver Metabolism After Alcohol Consumption
Levels of the amino acids alanine and proline in the liver were less in group A compared to group C (p < 0.002, Table 1). The liver proliferation process could explain this difference. The production of new hepatocytes can promote an acceleration of alcohol detoxication. The number of binuclear hepatocytes significantly increases in animals receiving alcohol, and the concentrations of amino acids negatively correlate with the number of binuclear hepatocytes [23]. Therefore, the consumption of amino acids could be increased during liver proliferation. Moreover, medical treatment with a high content of amino acids, such as branched-chain amino acids (BCAA), has positive effects on patients with liver disease [24].
The liver contains many detoxification agents, for instance, glutathione which was again less in group A than in group C in this study (p < 0.002, Table 1). Glutathione can bind to toxic compounds and neutralize them [25]. Additionally, selective exhaustion of the mitochondrial glutathione pool represents an essential pathophysiological change, by up to 60%, in chronic alcoholic liver diseases [26].
4.2 Brain Metabolism After Alcohol Consumption
Overall, the impact of alcohol on the brain is weaker than on the liver. Only three metabolites exhibited differences (aspartate, glutamine, and taurine, p < 0.05, Table 1) between the experimental groups, but after Holm–Bonferroni correction, none of the metabolites was different between groups. However, this finding is in agreement with a previous study [9], in which the alcohol doses were lower, and no changes in brain metabolism could be found. The fact that alcohol affects brain metabolism to a lesser extent than liver metabolism can be associated with brain metabolic stability or the existence of the blood–brain barrier.
4.3 Serum Metabolism After Alcohol Consumption
Lactate (p < 0.001, Table 1) levels were statistically smaller in serum samples in the alcohol group than in the control group but not in the liver, which differs from the results of our previous study [15]. Lactate is synthesized primarily from pyruvate in glycolysis and transforms into pyruvate via gluconeogenesis. However, ethanol inhibits glucose degradation (in this case, glycolysis) [27]. A lower lactate content in group A could indicate that less lactate is produced than consumed. Nicholas et al. [9] demonstrated decreased level of lactate and alanine in the liver and serum. These findings are in disagreement with a commonly accepted concept that lactate levels increase in blood under the impact of ethanol. Possibly, the inconsistency of these viewpoints is based on decreasing lactate levels in both liver and serum due to the futile cycling of pyruvate in the liver. Furthermore, it cannot be excluded that it is related to the duration of alcohol exposure and the energetic component of alcohol.
The concentrations of methionine (change on the verge of statistical significance, p = 0.019) and betaine were smaller in group A than in group C; they serve, for example, as sources of methyl groups for choline synthesis. Choline deficiency can cause nonalcoholic fatty liver diseases [28]. Moreover, methionine-choline deficiency diet also causes nonalcoholic fatty liver diseases [29]. In contrast, the addition of methionine [30] and betaine [31] to diet brings about improvements in liver diseases. However, the choline content is not different between groups here (p = 0.75).
The content of acetic acid and formic acid was higher in group A compared to group C. Acetic acid is an intermediate product of ethanol degradation, which can be released from the liver into the blood when alcohol has not been completely neutralized. Thus, some ethanol degradation products enter the bloodstream and can be delivered to other organs. The same may apply to formic acid.
4.4 Comparison of Metabolic Changes in the Liver, Brain, and Serum
The main advantage of metabolomic analysis based on blood samples is the minimally invasive blood sampling procedure. However, it is not apparent that blood can provide correct information about one particular tissue, ignoring all others. Blood interacts with other organs and can reflect the state of the entire body.
No unidirectional changes were detected between the liver and serum and brain and serum metabolic profile, and only three metabolites (glucose, betaine, and isoleucine) were correlated between serum and liver. Therefore, PLS-DA was performed to assess the variance between tissues and serum metabolic response to alcohol exposure. PLS-DA reduces the number of initial variables, and it is often used in metabolomics to maximize the separation between groups [32] and assess overall response to the experimental conditions. Both liver and serum were effectively separated by alcohol exposure factor in the axes Y2, in contrast to the brain. Here, too, the brain was affected less than the liver and serum. The similar tendency observed in the liver and serum can be explained by the common trend in differences within several metabolites' levels between group A and group C (e.g., alanine, creatine, valine, and isoleucine). The difference is significant at p < 0.05 (Table 1).
While the entire organism suffers from a toxic load under alcohol consumption, the toxic load and resistance are different for various organs. The liver suffers from numerous toxic effects, but it can recover much faster than, for example, the brain, which is subjected less to the toxic influence but has a negligible possibility to recover.
4.5 Limitations and Future Prospects
The metabolic content detected here was evaluated only by 1H NMR spectroscopy in vitro. Other nuclei, e.g., 13C can be used to investigate the transformation of pyruvate to lactate, the Krebs cycle, or selectively study the metabolites related to the ethanol consumption using, e.g., 13C-acetate [33]; however, isotope enrichment is desirable to obtain a high signal-to-noise ratio. Besides, a comparison with widely used methods such as histology, serological analysis, or measurement of gene expression as well as to the MR spectroscopy in vivo would be advantageous. All of these can reflect metabolic content and its changes and vice versa. Moreover, possibly, spectroscopy can detect changes that occurred before the tissue transformation or before the enzyme concentrations had changed. Thus, the combination of both metabolomic and conventional approaches is necessary for further studies. The quantitative metabolic information is presented here in arbitrary units, as the concentration of the metabolites was normalized by the sum of identified metabolites for each animal. Using this approach, we can detect the biological differences between the groups; however, adding a reference chemical compound such as DSS or TSS to each sample would provide absolute metabolite concentrations. Only three biological tissues (liver, brain, and blood) were investigated in this study. Other organs and body fluids, such as the heart, skeletal muscle, urine, etc., may also be affected by alcohol and could be studied to view the broader picture of alcohol impact or any other toxic load on the metabolism.
5 Conclusions
NMR spectroscopy in vitro can determine the metabolic differences between alcohol and control groups in liver and serum extracts. No metabolic differences between alcohol and control group in the brain were detected.
Data Availability
The data is available under request from a corresponding author.
Abbreviations
- ANCOVA:
-
Analysis of covariance
- BMRB:
-
Biological magnetic resonance bank
- BCAA:
-
Branched-chain amino acids
- DSS:
-
Sodium trimethyl silyl propane sulfonate
- HMDB:
-
Human metabolome database
- ICG SB RAS:
-
Institute of Cytology and Genetics of the Siberian Branch of the Russian Academy
- NMR:
-
Nuclear magnetic resonance
- PLS-DA:
-
Partial least-squares discriminant analysis
- SPF:
-
Specific pathogen-free
- TSS:
-
3-(Trimethylsilyl)-propionic acid sodium salt
References
C.S. Lieber, Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 34(1), 9–19 (2004)
I. Novo-Veleiro, L. Alvela-Suárez, A.-J. Chamorro, R. González-Sarmiento, F.-J. Laso, M. Marcos, Alcoholic liver disease and hepatitis C virus infection. World J Gastroenterol 22(4), 1411–1420 (2016)
J.L. Peng, M.P. Patel, B. McGee, T. Liang, K. Chandler, S. Tayarachakul et al., Management of alcohol misuse in patients with liver diseases. J Investig Med Off Publ Am Fed Clin Res 65(3), 673–680 (2017)
A. White, What happened? alcohol, memory blackouts, and the brain. Alcohol Res Health J Natl Inst Alcohol Abuse Alcohol 27(2), 186–196 (2003)
T. Julian, N. Glascow, R. Syeed, P. Zis, Alcohol-related peripheral neuropathy: a systematic review and meta-analysis. J Neurol 266(12), 2907–2919 (2019)
C. Cheng, C.-L. Huang, C.-J. Tsai, P.-H. Chou, C.-C. Lin, C.-K. Chang, Alcohol-related dementia: a systemic review of epidemiological studies. Psychosomatics 58(4), 331–342 (2017)
K. Nixon, Alcohol and adult neurogenesis: Roles in neurodegeneration and recovery in chronic alcoholism. Hippocampus 16(3), 287–295 (2006)
J. Lian, W. Liu, S. Hao, Y. Guo, H. Huang, D. Chen et al., A serum metabonomic study on the difference between alcohol- and HBV-induced liver cirrhosis by ultraperformance liquid chromatography coupled to mass spectrometry plus quadrupole time-of-flight mass spectrometry. Chin Med J (Engl). 124(9), 1367–1373 (2011)
P.C. Nicholas, D. Kim, F.T. Crews, J.M. Macdonald, 1H NMR-based metabolomic analysis of liver, serum, and brain following ethanol administration in rats. Chem Res Toxicol. 21(2), 408–420 (2008)
B.U. Bradford, T.M. O’Connell, J. Han, O. Kosyk, S. Shymonyak, P.K. Ross et al., Metabolomic profiling of a modified alcohol liquid diet model for liver injury in the mouse uncovers new markers of disease. Toxicol Appl Pharmacol 232(2), 236–243 (2008)
H.G. Gika, C. Ji, G.A. Theodoridis, F. Michopoulos, N. Kaplowitz, I.D. Wilson, Investigation of chronic alcohol consumption in rodents via UHPLC-MS based metabolite profiling. J Chromatogr A 12(1259), 128–137 (2012)
H.G. Gika, I.D. Wilson, Global metabolic profiling for the study of alcohol-related disorders. Bioanalysis 6(1), 59–77 (2014)
N. Loftus, A. Barnes, S. Ashton, F. Michopoulos, G. Theodoridis, I. Wilson et al., Metabonomic investigation of liver profiles of nonpolar metabolites obtained from alcohol-dosed rats and mice using high mass accuracy MSn analysis. J Proteome Res 10(2), 705–713 (2011)
C. Torruellas, S.W. French, V. Medici, Diagnosis of alcoholic liver disease. World J Gastroenterol WJG 20(33), 11684–11699 (2014)
M.S. Krestina, O.B. Shevelev, I.V. Koptyug, L.A. Gerlinskaya, S.E. Peltek, A.E. Akulov, Opportunities of high-technology phenotyping by the NMR spectroscopy method by example of the metabolic response of the rat liver to the effect of alcohol and Reishi. Russ J Genet Appl Res. 6(4), 485–490 (2016)
J.A. Obernier, T.W. Bouldin, F.T. Crews, Binge ethanol exposure in adult rats causes necrotic cell death. Alcohol Clin Exp Res 26(4), 547–557 (2002)
N. Lutz, J.V. Sweedler, R. Wevers (eds.), Methodologies for Metabolomics: Experimental Strategies and Techniques (Cambridge University Press, New York, NY, 2013), p. 627
N.W. Lutz, E. Béraud, P.J. Cozzone, Metabolomic analysis of rat brain by high resolution nuclear magnetic resonance spectroscopy of tissue extracts. J Vis Exp JoVE. 2014. https://doi.org/10.3791/51829
Y. Masuo, T. Imai, J. Shibato, M. Hirano, O.A.H. Jones, M.L. Maguire et al., Omic analyses unravels global molecular changes in the brain and liver of a rat model for chronic Sake (Japanese alcoholic beverage) intake. Electrophoresis 30(8), 1259–1275 (2009)
N. Psychogios, D.D. Hau, J. Peng, A.C. Guo, R. Mandal, S. Bouatra et al., The human serum metabolome. PLoS ONE 6(2), e16957 (2011)
P.S. Gromski, H. Muhamadali, D.I. Ellis, Y. Xu, E. Correa, M.L. Turner et al., A tutorial review: metabolomics and partial least squares-discriminant analysis–a marriage of convenience or a shotgun wedding. Anal Chim Acta. 16(879), 10–23 (2015)
R.G. Brereton, G.R. Lloyd, Partial least squares discriminant analysis: taking the magic away. J Chemom. 28(4), 213–225 (2014)
O.B. Shevelev, A.E. Akulov, A.S. Dotsenko, G.V. Kontsevaya, M.A. Zolotykh, L.A. Gerlinskaya et al., Neurometabolic effect of Altaian fungus Ganoderma lucidum (Reishi mushroom) in rats under moderate alcohol consumption. Alcohol Clin Exp Res 39(7), 1128–1136 (2015)
K. Tajiri, Y. Shimizu, Branched-chain amino acids in liver diseases. World J Gastroenterol WJG 19(43), 7620–7629 (2013)
L. Yuan, N. Kaplowitz, Glutathione in liver diseases and hepatotoxicity. Mol Aspects Med 30(1–2), 29–41 (2009)
J.C. Fernández-Checa, A. Colell, C. Garcı́a-Ruiz, S-Adenosyl-l-methionine and mitochondrial reduced glutathione depletion in alcoholic liver disease. Alcohol 27(3), 179–183 (2002)
S.Q. Siler, R.A. Neese, M.P. Christiansen, M.K. Hellerstein, The inhibition of gluconeogenesis following alcohol in humans. Am J Physiol-Endocrinol Metab. 275(5), E897–E907 (1998)
K.D. Corbin, S.H. Zeisel, Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr Opin Gastroenterol 28(2), 159–165 (2012)
É. Marcolin, L.F. Forgiarini, J. Tieppo, A.S. Dias, L.A.R. de Freitas, N.P. Marroni, Methionine- and choline-deficient diet induces hepatic changes characteristic of non-alcoholic steatohepatitis. Arq Gastroenterol 48(1), 72–79 (2011)
A. Kumar, R. Pathak, H.A. Palfrey, K.P. Stone, T.W. Gettys, S.N. Murthy, High levels of dietary methionine improves sitagliptin-induced hepatotoxicity by attenuating oxidative stress in hypercholesterolemic rats. Nutr Metab. 17(1), 2 (2020)
E. Kathirvel, K. Morgan, G. Nandgiri, B.C. Sandoval, M.A. Caudill, T. Bottiglieri et al., Betaine improves nonalcoholic fatty liver and associated hepatic insulin resistance: a potential mechanism for hepatoprotection by betaine. Am J Physiol-Gastrointest Liver Physiol 299(5), G1068–G1077 (2010)
A.A. Seryapina, O.B. Shevelev, M.P. Moshkin, A.L. Markel, A.E. Akulov, Stress-sensitive arterial hypertension, haemodynamic changes and brain metabolites in hypertensive ISIAH rats: MRI investigation. Exp Physiol 102(5), 523–532 (2017)
L. Jiang, B.I. Gulanski, H.M. De Feyter, S.A. Weinzimer, B. Pittman, E. Guidone et al., Increased brain uptake and oxidation of acetate in heavy drinkers. J Clin Invest 123(4), 1605–1614 (2013)
Funding
Open Access funding enabled and organized by Projekt DEAL. The animals and alcohol modeling were supplied through the state-financed project no. 0259-2019-0004-C01. 1H NMR spectroscopy and sample preparation was supported by the Russian Foundation for Basic Research (grant # 19-53-12013). Besides, we acknowledge the financial and intellectual support by the Research Training Group “Materials4Brain” (GRK2154; P2).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflicts of interest.
Ethics Approval
All procedures complied with the Directive of 22 September 2010 (2010/63/EU) of the European Parliament and with the Guide for the Care and Use of Laboratory Animals (Washington, DC, USA) and were approved by the Bioethical Committee of Institute of Cytology and Genetics Siberian Branch of Russian Academy of Sciences (SB RAS) in Novosibirsk, Russia. All efforts were made to minimize the number of animals used and their suffering.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pravdivtseva, M.S., Shevelev, O.B., Yanshole, V.V. et al. In Vitro 1H NMR Metabolic Profiles of Liver, Brain, and Serum in Rats After Chronic Consumption of Alcohol. Appl Magn Reson 52, 661–675 (2021). https://doi.org/10.1007/s00723-021-01338-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-021-01338-x