
Abstract
Quantitative nuclear magnetic resonance (qNMR) is considered as a powerful tool for multicomponent mixture analysis as well as for the purity determination of single compounds. Special attention is currently paid to the training of operators and study directors involved in qNMR testing. To assure that only qualified personnel are used for sample preparation at our GxP-accredited laboratory, weighing test was proposed. Sixteen participants performed six-fold weighing of the binary mixture of dibutylated hydroxytoluene (BHT) and 1,2,4,5-tetrachloro-3-nitrobenzene (TCNB). To evaluate the quality of data analysis, all spectra were evaluated manually by a qNMR expert and using in-house developed automated routine. The results revealed that mean values are comparable and both evaluation approaches are free of systematic error. However, automated evaluation resulted in an approximately 20% increase in precision. The same findings were revealed for qNMR analysis of 32 compounds used in pharmaceutical industry. Weighing test by six-fold determination in binary mixtures and automated qNMR methodology can be recommended as efficient tools for evaluating staff proficiency. The automated qNMR method significantly increases throughput and precision of qNMR for routine measurements and extends application scope of qNMR.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Quantitative nuclear magnetic resonance (qNMR) is an important method to assess the purity of organic compounds as well as to perform multicomponent analysis of complex mixtures. The application range of qNMR nowadays varies from small organic molecules to large proteins [1,2,3,4,5].
Following discussions within the NMR community it can be concluded that special attention is currently paid on optimization of the whole qNMR workflow. In this regard, standardized tools to estimate performance of both qNMR operators (laboratory staff) and data analysts have to be developed. This is important for all laboratories including GMP/GLP accredited ones. In the later case thorough documentation of each analysis step and internal personal training are also mandatory. These regulations are required to ensure that bio/pharmaceutical products are safe, meet their intended use, and adhere to quality processes during manufacturing, control, storage, and distribution.
In this study, we present one possible procedure to evaluate the precision and accuracy of sample preparation by performing six-fold weighing of binary mixtures. Our idea contradicts with the usual practice, where the weighing is used to validate the results of qNMR. However, since NMR signal intensity is directly proportional only to the number of nuclei of the corresponding functional group, this method can be also considered as an ab initio methodology. As the model system the mixture of 2,6-di-tert-butyl-4-methylphenol (BHT) and 1,2,4,5-tetrachloro-3-nitrobenzene (TCNB) was selected.
On the other side, data analysis is another crucial part of the qNMR workflow. A tool for automated integration in qNMR analysis has been recently developed, which helps to prevent transfer errors during data evaluation, identify spectral artefacts, and enables high throughput of the whole analysis workflow [6,7,8]. This routine also verifies preprocessing and integration steps in a fully automated manner. The approach was successfully tested on binary mixtures of qNMR internal standards having simple spectra, as well as complex multiplet patterns of pharmaceutical products [6,7,8]. It was hypothesized that our user-independent procedure can be used to check the quality of quantitative results. All accumulated data for binary mixtures therefore were evaluated manually by a qNMR expert and the above-mentioned automated routine. Moreover, we compare performance of manual and automated integration on the qNMR purity determination of 32 authentic compounds used in pharmaceutical industry. This paper summarizes the main outcomes of this study.
2 Experimental
2.1 Samples and Sample Preparation
Proficiency test was performed at an GxP accredited laboratory Spectral Service AG (Cologne, Germany) in Dec 2019. In particular, nine technicians and seven study directors performed sixfold weighing of binary mixtures (six replicates) of BHT and TCNB. These two substances were purchased from Sigma Aldrich (St. Louis, Missouri, USA), where they were certified by gas chromatography. TCNB was additionally certified in our laboratory by six-fold NMR determination; the purity was found to be 99.8 ± 0.1%. Accurately weighed quantities of about 15 mg of the substance (BHT) to be analysed and the reference material (TCNB) were dissolved in 0.7 mL of DMSO-d6. Each participant performed weighing in the same day. A Mettler Toledo XPE205DR balance (Greifensee, Switzerland) with repeatability of 0.015 mg for 1 g check weight was used as the balance (operating rang 0.001 to 10 g). The balance is located in air-conditioned room. The test solutions were mixed using a vortex mixer and/or an orbital shaker. The complete solubility of the solid material was checked visually. An aliquot of each solution was transferred to a NMR tube for quantitative 1H-NMR measurement. The 1H-NMR experiments were performed under the same operating conditions within 2 h after sample preparation.
Moreover, 32 authentic compounds used in pharmaceutical industry with molecular weight between 80 and 600 g/mol were also evaluated. The examples of authentic samples are bisabolols, ibuprofen, 1-bromo-4-methylamino-anthraquinone, 1-naphthol, benzoquinones, 4-fluoro-2-methoxy-5-nitroaniline and more complex ones. They were routine samples received for qNMR purity testing in our laboratory. CDCl3, D2O, DMSO-d6, and MeOD were used as solvents. Appropriate internal standard was chosen from the following list: thymol, nicotininic acid amid, 1,2,4,5-tetrachloro-3-nitrobenzene, 3,6-dichloro-2-methoxybenzoic acid, 1,2,4,5-tetramethylbenzene, benzylbenzoate (BBE), 4-dimethylaminobenzoic acid ethyl ester, 4-(trifluoromethyl)-benzonitrile, maleic acid, dimethyl sulfone. The purity of test items was calculated as the average value based on two separate qNMR measurements.
2.2 1H NMR Measurements and Preprocessing
NMR measurements were performed on Bruker Avance III 500/600 MHz spectrometers (Bruker Biospin, Rheinstetten, Germany) with BBO Prodigy cryo/QNP cryo probes equipped with Bruker Automatic Sample Changer (B-ACS 120) at room temperature. The standard acquisition parameters for weighing test were: 1D pulse sequence with 300 flip angle, relaxation delay (D1 = 40 s.), time domain (TD) 128 k data points, number of scans (NS) 16, acquisition time (AQ) 3.2727999 s., transmitter (frequency) offset position (O1P) 6.175 ppm, spectral width (SW) 19.9836 ppm.
T1 relaxation times were determined using inversion-recovery experiments. For BHT, the values were equal to 1.07, 1.53, 1.23 and 0.56 s for CH (aromatic), OH, CH3 and t-butyl functional groups, respectively. For the aromatic proton of TCNB, the T1 time was found to be 8.4 s. Therefore, relaxation delay of D1 = 40 s. was enough for quantitative studies.
All NMR spectra were phased and baseline-corrected using Topspin 3.5 (Bruker Biospin, Rheinstetten, Germany). A line broadening (LB) of 0.3 Hz was applied before FT.
2.3 Data Evaluation
The signals of aromatic protons (δ 6.78 ppm for BHT and δ 8.47 ppm for TCNB) were selected for quantification. Moreover, the BHT signals at δ 1.30 ppm and δ 2.15 ppm were also evaluated for comparison.
For automated NMR spectra evaluation, a macro was written within the NMR Bruker software, which connects previously developed Matlab script and the Topspin environment [6,7,8]. For spectra evaluation, only integral borders and the center of the signals have to be defined manually by an operator. The output consists of average BHT purity, average integral values along with standard deviation, and number of integrals that were considered for calculations. The number of integrals varied between 80 and 300. The automated routine takes about 5 s per sample. Approximately 30 min is required for manual spectral evaluation.
Successive integration is performed by continuous adding one data point from both sides of the signal “core” (40 Hz) till the integration borders are reached. A typical feature of an NMR signal that its intensity diminishes quickly from its center in a 40–50 Hz window was considered. Integration is based on the trapezoidal method with unit spacing. Two integration strategies were applied to each peak. First, successive integration is performed by continuous adding one data point from both sides of the signal “core” till the signal boarders are reached. In the second case, consecutive integration is performed in two opposite directions from the “core” signal and two resulting parts were combined in one profile. The resulted evolving profiles were normalized to 100%. The average value from all integrals above 99.9% was used to assess BHT concentration. The script was adjusted when another number of BHT nuclei generated the signal. Our routine checks the presence of phase and baseline distortions as well as unrelated resonates in the analyte region. Further details can be found in our previous publications [6,7,8].
Principle component analysis (PCA) using SAISIR toolbox for Matlab (The MathWorks, Natick, USA) was used for data exploration [9].
t- and F- statistical test were used to evaluate significance of intercept of linear curves. In all cases, it was found to be insignificant and was set to zero. The tabulated values used to create Figs. 3, 4, 6 can be found in Supporting information (Table S1–S3).
3 Results and Discussion
3.1 Manual and Automated Evaluation of the Six-fold Weighing Experiment
As an example, 1H-NMR spectrum of binary BHT-TCNB mixture in DMSO-d6 is shown in Fig. 1. TCNB produces one signal for the aromatic proton at 8.47 ppm. The peaks of BHT at δ 6.87, δ 6.65, δ 2.15 and δ 1.30 ppm originated from two, one, three and eighteen protons, respectively. These two compounds are often used in routine qNMR work due to their solubility in many organic solvents. Both compounds have an aromatic signal, which do not overlap with resonances of matrix compounds in many cases. BHT has four peaks originated from different number of protons, which is predestinated to study linearity of NMR system.

Typical 1H-NMR spectrum of the BHT–TCNB mixture in DMSO-d6. TCNB produces one signal for the aromatic proton at 8.47 ppm. BHT shows peaks at δ 6.87, δ 6.65, δ 2.15 and δ 1.30 ppm
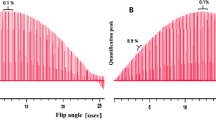
Due to low enough relaxation time values, absence of disturbing signals (i.e., DMSO signal at δ 2.5 ppm), aromatic signals of both substances were first selected for calculations. Figure 2a, b presents the performance of the automated integration routines applied to these peaks [6, 7]. Such integration profiles are the marker of accurately performed preprocessing and integration.

NMR signal of TCNB (a) and BHT (b) with two integration profiles (lower plots). Color horizontal lines restrict the typical signal area without 13C satellites. The detailed description of the integration procedure can be found in [6] (color figure online)
Using both manual integration by a qNMR expert and our automated procedure, 96 qNMR spectra (sixfold determination performed by 16 people) were evaluated and the results are summarized by mean value, standard deviation and relative standard deviation (RSD) for each of 16 participants. The same concentration was used for all samples to avoid additional uncertainty of results. Figure 3 shows the relation between mean values obtained by manual evaluation and automated routine. A linear fit was applied to the data, and the fitted line had a slope of 1 and no y-axis offset. It means that the mean values are similar and both evaluation approaches are free of systematic error. However, looking on the slope of linear fit obtained for relative standard deviation values from each of six-fold determination, automated evaluation resulted in approximately 20% increase in precision (Fig. 4). This shows that qNMR uncertainty was reduced, the maximum RSD value among all participants in our weighing test was 0.35%. To investigate accuracy references methods/standardize reference materials are required, which was outside the scope of this work. However, loss of accuracy is not expected in this case.

Relation between mean values obtained by manual evaluation and automated routine. The results of laboratory staff (blue) and study directors (red) are marked with different colours. The regression curve fits all points for laboratory staff and study directors (color figure online)

Relation between RSD obtained by manual evaluation and automated routine. The results of technicians (blue) and study directors (red) are marked with different colours. The regression curve fits all points for laboratory staff and study directors (color figure online)
Both technicians and study directors participated in weighing. The difference in performance between these two groups was noticed in mean values and RSDs (Figs. 3, 4). Qualified technicians (laboratory staff) who perform the operation frequently weighed more precisely (smaller standard deviation, mean values are close to each other) than study directors. The same results were derived by principal component analysis (PCA) analysis of the results (Fig. S1). Moreover, the scores plot showed no differences between two evaluation procedures. Using transposed data matrix (participants in columns and six independent measurements in rows) it was observed that the third and the fourth weighing were the most stable ones (points C and D were located close to the origin of score plot). On the contrary, the first and the sixth values (points A and F on the score plot) deviated more from the corresponding mean value.
Only trained and instructed personnel are authorized to carry out the sample preparation for qNMR analysis under GxP conditions. In this weighing test, a staff member is considered successfully trained if the mean BHT purity and corresponding RSD are within 99.7% ± 0.3% and ≤ 0.4%, respectively. The BHT purity according to the reference GC analysis was found to be 99.7%. Our weighing training has shown that all participants have the competence, which is required to ensure correct results in sample preparation. Another way for checking technician´s proficiency is to perform weighing of a given mass standard. This, however, is not a part of a routine qNMR workflow, which includes other steps such as dilution and transferring in a NMR tube. Therefore, such weighing test by six-fold determination in binary mixtures can be recommended as a tool for checking technicians’ proficiency involved in GxP studies.
3.2 qNMR on different BHT signals
Since our test item (BHT) produces several NMR signals, which can be used for qNMR evaluation, the automated routine was tested on two further cases (Fig. 1). Theoretically, if the excitation profile is uniform enough across the spectral window, and that the inter-scan delay is long enough to allow for complete relaxation of all nuclei, then all BHT resonances should be suitable for quantitation.
The mean values and corresponding RSDs depend strongly on the signal used for qNMR (Fig. 5, S3). Peak areas of aliphatic protons cannot be fully integrated because of broadness of the signal δ1.30 ppm and proximity of the signal at δ 2.15 ppm to DMSO signal. This results in underestimation of BHT content (Fig. 5). The signal at δ 1.3 ppm (18 protons) has a very broad halfwidth and the integral limits have to be set very wide to cover close to 100% of integral area. Unfortunately, this is impossible due to neighbour signals. Another signal at δ 2.15 ppm is close to the solvent signal, and that again prevents accurate integration.

Mean values of six-fold determinations using different BHT signals and the aromatic peak of TCNB
RSDs values showed similar tendency: the biggest average RSD of 0.24% was obtained for the signal at δ 2.15 ppm, followed by 0.21% at δ 1.30 ppm and 0.16% at δ 6.78 ppm (Fig. S3). The situation would be better if we take the sum of the three BHT integrals and divide it to the total number of protons (24). Therefore, our results have proved that only aromatic signal is appropriate for quantification. Thus, our automated routine can easily identify peaks most suitable for integration and identify not optimal evaluation conditions.
3.3 Application to Authentic Samples
Since the performance of weighing is assured, special tools should be developed to control NMR data analysis, in particular, manual preprocessing and peak integration steps. In this regard, our automated routine was used. Several studies directors routinely performed purity determination of authentic compounds used in pharmaceutical industry, where peak purity is not automatically given. For sample preparation, appropriate solvent as well as internal standard were selected (see experimental section). Peaks that carry the highest quality information (no peak overlap and the highest signal-to-noise ratio) were chosen for quantification. As an example, 1H-NMR spectrum of ibuprofen as well as integration profiles were shown in Figs. 6, 7. In most cases, single qNMR purity values obtained by automated and manual quantification routines for all samples showed good agreement (slope is 1.00, y- intercept is 0.0, and R2 is 1.0). This means that manual integration was performed correctly.

1H-NMR spectrum of ibuprofen in DMSO-d6. TCNB was used as internal standard. The resonances selected for integration are highlighted for active ingredient and internal standard as AI and IS, respectively. The purity was found to be 99.7%

Integration profiles obtained by the automated routine for ibuprofen
A large discrepancy was noticed between single determinations for one sample (two replicates for this sample marked in circle in Fig. 8). The outliers were not used for calculation of R2, slope and intercept. 1H-NMR spectrum of this compound was shown in Fig. 9. The low quality results can be explained by complex multiplet pattern and disturbing resonance at δ 3.34 ppm, which lowered the required integration border (Figs. 9, 10). A poor homogeneity can be an additional reason for the discrepancy. Due to big deviation, the two outliers are not at the same side of the diagonal line in Fig. 8. For such samples, even automated integration cannot provide accurate results. To handle such situations, other solvent and/or decoupling pulse sequences can be tried. The integration profiles for internal standard signals (TCNB and BBE) for both samples can be found in supporting information (Figs. S4, S5).

Relation between mean purity values obtained by manual evaluation and automated routine for double qNMR determinations of 32 authentic compounds used in pharmaceutical industry. Two replicates of one outlier (marked in circles) can be explained by the crowded NMR integration region and poor homogeneity. The outliers were not used for calculation of R2, slope and intercept

1H-NMR spectrum of a pharmaceutical ingredient in DMSO-d6. BBE was used as internal standard. The resonances selected for integration are highlighted for active ingredient and internal standard as AI and IS, respectively. The purity was found to be 93.9%

Integration profiles obtained by automated routine for the active ingredient depicted in Fig. 9
The results have shown that automated routine can be used to check the quality of manual data evaluation including phase- and baseline adjustment as well as integration. However, introduction of automated procedure instead of manual evaluation in routine qNMR practice results in higher throughput, safety and precision of qNMR analysis.
4 Conclusion
NMR is an appealing platform for conducting quantification studies on complex systems [1,2,3,4,5]. To ensure robustness and precision of the method, several practical guides for spectra acquisition and accurate processing were developed [10]. As NMR continues to carve a niche in highly regulated environments, guaranteeing that scientific personnel are adequately trained to perform all of the tasks associated with the analysis is critical.
In this paper, it was shown that the quality of sample preparation can be evaluated by performing a simple six-fold weighing of a binary system. In this regard, NMR is used as a a priori standard to estimate the quality of weighing. It is possible to extend the conclusions to more complex multi-component systems.
Moreover, our trials have shown that an automated analysis routine can be used to check the quality of data analysis. On the other side, this procedure can be recommended for routine qNMR analysis workflow. Besides our routine, several competing automated methods were presented for performing NMR peak integration without prior knowledge of compound structures and using an external calibration spectrum [11,12,13].
Binary mixture of BHT and TCNB was selected for this study. In the next step, a certified reference material of known content/purity can be used to evaluate not only the precision of the measurements, but also the accuracy of the determinations. Moreover, analysis of acquired data by different study directors (also at interlaboratory level) would be interesting.
We also plan to perform more extensive tests using broader data sets of authentic samples using reference methods. In this case, the accuracy of NMR methodology can be additionally justified. Furthermore, we plan to develop a procedure, which could automatically detect the most suitable signals of internal standard and test item for integration. These activities would make the qNMR automation process more robust and accurate.
References
B. Diehl, U. Holzgrabe, Y. Monakhova, T. Schönberger, J. Pharm. Biomed. Anal. 177, 112847 (2020)
W.C. Brooks, N.D. Paguigan, H.A. Raja, F.J. Moy, N.B. Cech, C.J. Pearce, N.H. Oberlies, Magn. Reason. Chem. 55(7), 670–676 (2017)
J.E. Melanson, M.P. Thibeault, B.B. Stocks, D.M. Leek, G. McRae, J. Meija, Anal. Bioanal. Chem. 410(26), 6719–6731 (2018)
T. Huang, W. Zhang, X. Dai, X. Zhang, C. Quan, H. Li, Y. Yang, Talanta 125, 94–101 (2014)
S. Mahajan, I.P. Singh, Magn. Reson. Chem. 51(2), 76–81 (2013)
Y.B. Monakhova, B.W.K. Diehl, Magn. Reason. Chem. 55(9), 813–820 (2017)
Y.B. Monakhova, B.W.K. Diehl, Magn. Reason. Chem. 55(11), 996–1005 (2017)
Y.B. Monakhova, B.W.K. Diehl, Magn. Reason. Chem. 57(2–3), 110–117 (2019)
C.B.Y. Cordella, D. Bertrand, Trends Anal. Chem. 54, 75–82 (2014)
T. Saito, S. Nakie, M. Kinoshita, T. Ihara, S. Kinugasa, A. Nomura, T. Maeda, Metrologia 41, 213–218 (2004)
X. Liu, M.X. Kolpak, J. Wu, G.C. Leo, Anal. Chem. 84, 6914–6918 (2012)
P.A. Hays, T. Schoenberger, Anal. Bioanal. Chem. 406, 7397–7400 (2014)
E. Alm, T. Slagbrand, K.M. Aberg, E. Wahlström, I. Gustafsson, J. Lindberg, Anal. Bioanal. Chem. 403, 443–455 (2012)
Acknowledgements
We warmly thank Mike Bernstein for his useful suggestions. Y. Monakhova acknowledges support of the Russian Science Foundation (project 18-73-10009).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Monakhova, Y.B., Diehl, B.W.K. A Step Towards Optimization of the qNMR Workflow: Proficiency Testing Exercise at an GxP-Accredited Laboratory. Appl Magn Reson 52, 581–593 (2021). https://doi.org/10.1007/s00723-021-01324-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-021-01324-3