
Abstract
A recent paper on solid-state 13C nuclear magnetic resonance in graphitic materials reports on the difficulties with the interpretation of the spectra and presents alternative methods to obtain informative experimental data. Therein, special emphasis is placed on the role of dipole–dipole interactions in the case of 13C-enriched samples and on the assignment of the 13C spectral components beyond the common “graphitic range”. Here, we show that the consideration of the experimental conditions, namely, the speed of magic angle spinning, plays a more important role in the data interpretation. Further, we provide the experimental and theoretical evidences that the appearance of the 13C resonances shifted upfield from the typical sp2-hybridized carbon range is not surprising, but rather exhibits a characteristic feature of the graphitic materials with specific morphology. Finally, we show that analysis of the anisotropy of the chemical shift tensor is informative even in the case of very broad 13C spectra and supports the microscopy observations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A recent paper by Freitas reports on difficulties with the interpretation of solid-state 13C nuclear magnetic resonance (NMR) spectra in graphitic materials with special emphasis on the case of 13C-enriched samples [1]. The author doubts the fitting models used in our recent publication [2], where we reported the 13C magic angle spinning (MAS) NMR spectra of fine-grained graphitic materials in order to understand the effect of hot pressing on the structural properties of the materials. That work is one of the series of our publications on the studies of thermal [2, 3], chemical [4, 5] and electrochemical [6] treatments of graphitic materials with natural and enriched 13C isotope abundance. In these works, we applied solid-state NMR in order to observe changes in local atomic environments and in the structural properties of the materials. In most cases, except for fluorinated graphtes [4] and lithium-intercalated graphitic electrodes [6], 13C MAS NMR spectra demonstrated a broad asymmetric upfield-shifted signal. In order to extract the spectral parameters from the 13C NMR spectra, we applied a fitting model based on chemical shift anisotropy (CSA) interactions. In Ref. [1] Freitas suggests that dipole–dipole interactions play a more important role in fitting the 13C MAS NMR spectra than CSA interactions, particularly, when one deals with a higher concentration of NMR active spins upon 13C isotope enrichment. Moreover, the author criticizes [1] the assignment of the upfield spectral components in our works on 13C enriched graphitic materials [2, 3], which appear apart of the common “graphitic range”. We fully disagree with his statements and herein summarize the points of discussion as follows:
-
1.
For fitting 13C MAS NMR spectra, dipole–dipole interactions can be taken in consideration only at certain circumstances;
-
2.
The upfield 13C NMR signal can arise from graphitic carbon, as shown by some theoretical and experimental works;
-
3.
CSA MAS fitting model is best suited to describe the 13C MAS NMR spectra of 13C enriched graphitic materials in our works and even plays a crucial role for data interpretation.
1 Role of 13C–13C Homonuclear Dipolar Interactions in Fitting the 13C MAS NMR Spectra
In Ref. [1] Freitas states that “the 13C–13C homonuclear dipolar coupling also starts to contribute to the broadening (which can be difficult to overcome completely by MAS, depending on the spinning rate), to an extent that also increases with the enrichment factor”. A fast MAS NMR technique is known to average out the homonuclear dipolar coupling and narrow the line width. A simple calculation of the strength of dipolar interaction of a 13C–13C spin pair in a graphene sheet with a distance of 1.42 Å between nearest neighbors renders a value of ca. 4 kHz. It is evident that the contribution from the homonuclear dipolar interaction is fully averaged out in the 13C MAS NMR spectra measured with a spinning speed of 20 kHz, as used in our work [2]. In the case of a high 13C isotope enrichment of a graphitic material, where the occurrence of 13C–13C spin pairs is more frequent, we need to consider a cluster consisting of sp2-hybridized 13C atom, connected to three others carbons. When this cluster contains in average two 13C–13C spin pairs (that corresponds to 13C-enrichment of 65% as in our materials), MAS speed of 20 kHz is sufficient to average the homonuclear dipolar coupling.
An experimental evidence against the consideration of the homonuclear dipolar interactions for the 13C line shape has been provided in our recent publications [3, 4, 6]. In these works, we demonstrated that introduction of adatoms to 13C-enriched graphitic materials by a chemical or electrochemical way results in a drastic narrowing of the experimental 13C MAS NMR spectra. In Table 1, we summarize the line width parameters, when the graphitic materials are modified with heteroatoms. The results show that the addition into graphitic space of heteroatoms in small concentrations causes the significant decrease of the line width. This seems surprising, particularly considering the facts that the systems become more complex (heterogeneous) after modification and that all heteroatoms have NMR-active isotopes (79Br, 19F, 7Li and 23Na) with a relatively large γ and a high natural abundance.
If line width was determined by dipolar interactions, this would induce an additional broadening mechanism, which could result in a remarkable line broadening. However, our experimental data show the opposite trend suggesting another origin of line broadening in pristine materials, which is discussed below.
2 Upfield 13C NMR Signals in 13C Enriched Fine-Grained Graphitic Materials
In our studies [2, 3, 6] we reported an occurrence of the 13C spectral components in the ranges of 60–77 ppm and 27–37 ppm and assigned them to sp2-hybridized carbons undergone to specific interactions. In Ref. [1] Freitas questions whether “these spectral components are indeed true NMR signals or if they are artifacts resulting from the use of an inappropriate fitting model”. In addition, the author claims that “there is again no support from either previous experimental or computational studies to explain such an enormous isotropic shift of the resonance lines from the usual shift range expected from sp2 carbons (~ 100–130 ppm)”.
The spectral components found in our works indeed find little support in literature, because the available literature is poor and controversial particularly for the high-field region. The spectra are poorly resolved even at high MAS speed and diverse strongly depending on graphite precursor, synthesis conditions and the presence of defects. However, such “enormous isotropic shifts” have been encountered in the previous publications, for example, in the work reported by Freitas et al. some time ago [7]. The 13C shift of ca. 60 ppm was found for unmilled graphite and attributed to the effects of magnetic susceptibility. The changes of the shift and line width were found to correlate with the average crystallite sizes for milled graphites, where the decrease of the density of ring currents is expected. Moreover, a low surface area and the oxidation temperature of 610 °C in the unmilled graphite in Ref. [7] were similar to the parameters reported for our 13C graphites in Ref. [3].
In Ref. [1] Freitas claims that “previous reports have attributed signals at this chemical shift range to sp3 carbons in structurally disordered regions” citing the paper by Golzan et al. [8]. However, the author ignores for some reason the extension of the discussion, where Golzan et al. suggest that “The existence of a fraction of atoms, which have the compressed graphite configuration, is also consistent with the observation of a 66 ppm shift. Such configuration would have shifts intermediate between those of graphite and diamond with a spread of shifts…”.
The component at 27–37 ppm (even higher-field shifted) has been found only in thermally treated 13C graphitic materials [2, 3]. To increase the credibility of fit, we provided an additional two-step justification of the presence of the high-field peak (see Fig. 1). In Fig. 1a, we applied fit using a single component consisting of a central line and two spinning side bands (ssb) due to MAS denoted by asterisks. It is obviously that this fit is not satisfactory and one needs at least one more component upfield shifted, which can better describe the experimentally observed ssb. The second justification is based on a difference spectrum as shown in Fig. 1b, which represents a subtraction of the spectra of starting and treated 13C-samples scaled to have the same integral area. This shows a meaningful difference with a negative intensity at 30 ppm and positive intensities at 70 ppm and 122 ppm. This unambiguously demonstrates the appearance of a new component at 30 ppm and decreasing the other signals, respectively, after temperature treatment.

Copyright Elsevier © 2017
a Fit (grey) of the 13C MAS NMR spectrum of treated 13C-sample (black) with a single CSA line showing a mismatch of the experimental and simulated spinning side-bands (ssb). Asterisks denote the experimental ssb, grey dashed arrows point out to simulation. An arrow points to a kink originating from a minor line at 120 ppm. b Difference spectrum (black line) of starting and treated 13C-samples scaled to have the same integral area. Sum of fit components shows a very good agreement with the experimental difference spectrum. Reprinted with permission from Koroteev et al. Carbon 161, 124 (2017).
Further, Freitas states that “there are many examples of the previous works dealing with the calculation of NMR-shielding parameters using density functional theory (DFT) in carbon materials exhibiting structural distortions and defects; none of them predicts the existence of NMR signals associated with sp2 carbons having chemical shifts around 30–70 ppm”. First, we would like to emphasize that the DFT calculations of NMR parameters in graphitic materials have to be considered with extreme care, because “important macroscopic fields—the de-magnetizing field and the Lorentz internal field—yield strong shifts of the shift tensor component over hundreds of ppm, depending on the sample shape” as stated in a review paper by Goze-Bac et al. [9]. However, similar ab initio calculations of the magnetic shielding inside and outside carbon nanotubes originating from electronic current densities induced by the application of an external magnetic field have been performed a decade ago [10, 11]. In Ref. [10] it has been shown that magnetic screening effect experienced by guest molecules inside the tubes can be quite large resulting in the nucleus-independent chemical shift (NICS) values of 30 to − 60 ppm with larger tubes yielding more negative values. Further, a diamagnetic shift of 21 ppm has been predicted for the inner tube due to the outer one in a double-wall nanotube system [10]. This value coincided with the 13C shift measured directly on the 13C isotope enriched inner tube [12]. Moreover, it has been reported in Ref. [10] that in a double-wall nanotube, the NICS value inside the innermost tube is the sum of the two single-wall nanotube values, thus manifesting the superposition principle.
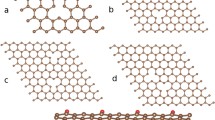
All these findings give us the full confidence that the upfield shifts observed in our work arise from graphitic carbon atoms, which experience a screening of the externally applied magnetic fields when located in a host superstructure. When we indeed look at the HRTEM images of graphitic particles presented in Fig. 2, we find curved/folded particles, which consist of several graphitic layers indicated by red lines (Fig. 2a). These particles can be imagined as few-wall carbon nanotubes of a large diameter, in those the inner tubes experience a magnetic screening causing the strong upfield 13C shift. In the thermally treated sample (Fig. 2b), morphology changes demonstrating closed conical-like tips, indicated by a yellow line, and more walls in the tubular system. Thus, the superposition principle mentioned above can be applied to this system to explain the signals in the range of 27–37 ppm. Thus, “the hypothesis of strong shielding caused by structural distortions in sp2 carbons even in the case of the well-graphitized samples annealed at 1500 °C—which is quite odd” on the opinion of the author of Ref. [1] has found a solid support in earlier theoretical studies [10, 11].

Copyright Elsevier © 2017 (color figure online)
HRTEM images of a starting (13C) and b hot pressing treated (13C-1200) graphitic materials. Red lines denote graphitic fragments, which can be imagined as multi-wall carbon nanotubes with a very large diameter d∞. Yellow line labels graphitic fragment, which can correspond to the upfield shifted spectral component, where magnetic screening is the strongest due to superposition principle. c Sketch of the structure and morphology of main kinds of graphitic particles present in 13C and 13C-1200. Adopted with permission from Koroteev et al. Carbon 161, 124 (2017).
At this point, we would like to comment the erroneous understanding of the author of Ref. [1] of the effects of magnetic susceptibility on 13C spectra. Freitas claims that “these magnetic susceptibility effects are expected to increase with the improvement in structural order in the graphitic lattice” [1] and exemplifies this statement through his study of milled graphites [7]. It is well known that magnetic susceptibility is a complex quantity determined by the contributions of the atomic-core diamagnetism, the Pauli paramagnetism and Landau diamagnetism of conduction electrons, the orbital diamagnetism, the Curie paramagnetism, and the orbital paramagnetic van Vleck contribution [13]. In milled graphites reported in [7], the diamagnetic susceptibility of graphene planes of a finite size was primarily controlled by the charge-carrier concentration related to the plane sizes (Landau diamagnetism). The contribution of ring currents has been found to be small or negligible (< 10 ppm) as long as a single plane is considered [9]. In contrast, in nanosized graphites or quasi-2D-graphites the other contributions have to be taken into account, which are governed by edge geometry (armchair or zigzag), stacking order and structural defects. It is clear that these contributions have different magnitudes and signs, and thus the total value is determined by addition or mutual compensation of the individual susceptibility components. Therefore, unambiguous correlation of the magnetic effects with structural ordering of graphites is only possible for a set of similarly prepared samples, as done in Ref. [7], and is not applicable for 13C fine-grained graphitic materials those structure and morphology are strongly affected as a result of thermal and chemical treatments [2, 3, 6]. However, the strong effects of locally anisotropic magnetic susceptibility have been indeed demonstrated in our materials, where the largest part of 13C spins located in well-ordered regions is quenched upon the action of demagnetizing fields. This part becomes visible if the diamagnetism is destroyed when lithium ions are intercalated into graphitic space and gradually disappears when diamagnetism is restored upon deintercalation [6].
3 Fitting Model for 13C MAS NMR Spectra
The next critic point raised by Freitas in Ref. [1] relates to the model we used to fit the NMR spectra in our work [2]. He asks whether “the 13C MAS NMR spectra of the 13C-enriched graphitic materials discussed in [2] perhaps could be fitted with components with more complex lineshapes (other than Gaussian/Lorentzian peaks), taking into account the existence of several combined contributions to the line broadening (e.g., distribution of isotropic chemical shifts, locally anisotropic magnetic susceptibility effects and residual 13C–13C homonuclear dipolar couplings)?”.
First of all, it would be interesting to understand what is the author’s meaning of “residual 13C–13C homonuclear dipolar couplings?”. It is well known that such effects are inherent to the strongly coupled spins, that is, with high γ and high isotope abundance, and can be applied to such nuclei as 1H, 19F and 31P. Taking into account that γ contributes to the strength of the dipolar interaction to a power of 2, the homonuclear dipolar coupling between 13C spins is 16 times weaker than between 1H, 14 times that between 19F, and almost three times than between 31P spins, without consideration of the 100% abundance of the latter.
In the following we discuss the contributions to the line broadening in our quite specific 13C graphitic materials and the choice of fitting model. It is well known in NMR spectroscopy that line broadening in solids arises from both homogeneous and inhomogeneous processes. Whereas the former is usually associated with dipolar broadening and can be narrowed by fast MAS, the latter arises from multiple structural sources producing different chemical shifts, and can be refocused in a spin-echo experiment. Indeed, for starting 13C fine-grained graphitic material, an inhomogeneous broadening mechanism has been revealed from slow decrease of the Hahn-echo intensity [6]. Most common sources for the latter are known to be inhomogeneity in the static field, a variation in bulk magnetic susceptibility within the sample, a distribution of isotropic chemical shifts, and a residual CSA resulting from incomplete motional narrowing. Indeed, macroscopic or microscopic non-homogeneities within the sample associated with structural irregularities, different phases and interfaces may introduce differences in the bulk magnetic susceptibility resulting in a slight line broadening. The detailed physical consideration of the effects of anisotropic bulk magnetic susceptibility for glassy and polycrystalline materials predicts the presence of the distributions of chemical shifts and hence line broadening, which is not spun out completely by MAS [14]. Moreover, in aromatic substances this effect is even stronger due to the large magnetic anisotropy of aromatic rings.
For the 13C graphitic materials, we suggest that the main contribution to line broadening is chemical shift dispersion. Heterogeneities in molecular packing, which are clearly visible in microscopy data, cause variations in the spatial arrangement of local molecular sources of magnetic susceptibility anisotropy giving rise to the distribution of chemical shifts. It is worth mentioning that well-ordered large graphitic particles are not visible in the spectra due to the effects of demagnetizing fields as discussed above and reported in our recent paper [6].
CSA MAS fitting model as implemented in DMFit software [15] includes all necessary parameters to describe the spectral lines, such as chemical shift, line width and the spectral area. While the line width comprises the information on inhomogeneous line broadening of ever origin, anisotropy of chemical shift can be characterized by fitting the ssb. The 13C MAS NMR spectra of 13C-enriched graphitic materials presented in [2] consist of two ssb and the inhomogeneously broadened central line, those asymmetric line shape allows to deconvolute the spectrum with several spectral components. We are confident that anisotropy is difficult to determine based on two ssb only. Therefore, it is clear that apparent trend as a function of the annealing temperature in [2] may lie within fit uncertainty. However, in following we summarized the anisotropy of the chemical shift tensor (δani) values for all spectral components of the 13C spectra reported in [2].
For the lines resonating at around 118, 70 and 30 ppm, the values of δani are found to be 85 ± 20, − 160 ± 20 and − 255 ± 45 ppm, respectively. Such large values allow us to conclude about sp2-hybridization state for all carbons and prove in addition that the line at 30 ppm has the largest δani and therefore cannot be assigned to sp3 carbon. Moreover, an obvious correlation of the parameters of chemical shift tensor, δiso and δani, helped us to additionally support our interpretation. Thus, the lowest value of 85 ± 20 ppm arises from graphitic nanoparticles observed in HRTEM (Fig. 2). Due to limited size (< 10 nm) they are not undergone to the strong effects of locally anisotropic magnetic susceptibility and therefore resonate in the chemical shift range expected for sp2 carbon (at 114 ppm). The value of 160 ± 20 ppm is typical for graphitic materials [9]. We attribute it to larger curved/folded particles, consisting of several graphitic layers, those inner layers experience a magnetic screening effects. Finally, δani of 255 ± 45 ppm, which is higher than that reported for graphites [9], is observed for the peak at ca. 30 ppm. This observation supports our suggestion that this component originates from strong bending of graphitic sheets after hot pressing (in other words, adjusting layers are coupled or fused as visible in HRTEM), which is marked by a yellow line in Fig. 2b.
It is important to state that the available NMR data does not allow to give a definitive “all-is-resolved” answer to the structural question under study. This appeals to the typical problem of solid-state NMR spectroscopy of “unfriendly” compounds, when the signals are so broad that it is impossible to validate the correctness of any interpretation from the NMR results alone. Credibility of our claims could in principle be cross-checked by an independent experiment, in which NMR sonde molecules confined in these closed graphene loops experience the same shift offset (50 ppm, 100 ppm). Our published results on (de-)lithiated 13C graphitic materials demonstrate a high-field 7Li shift of 10 ppm attributed to entrapped Li after delithiation [6] and provide proof of our interpretation. Considerably smaller 7Li shift than observed in 13C data can be due to less effective screening in those entrapment regions. Currently we have no ready-made solution how to inject guest semi-mobile molecules specifically into the regions, which undergone to maximal screening effects. Taking in consideration these difficulties, in our works on 13C graphites the NMR results have been interpreted in conjunction with the other characterization methods in order to get the comprehensive picture on these materials. Additional independent experiments for supporting our claims could be SIMPSON simulations addressing the influence of homonuclear 13C–13C interactions upon MAS spectra and field-dependent and/or spinning-speed dependent spectra accompanied by simulations for improved determination of CSA parameters and distributions of isotropic chemical shift components. Such experiments are however beyond the scope of the present comment and will be presented in future work.
Before summing up, we would like to point out some discrepancies attracted our attention in Ref. [1]. First, Freitas mentions that “the XPS results presented in [2] show the occurrence of contributions that are not completely consistent with the NMR results”. Indeed, deconvolution of a broad unresolved NMR line with several minor peaks is not unambiguous. Therefore it was reasonable to apply in [2] a so-called “chemical modeling”—a common approach to fit the spectra based on the information available from other methods. In our paper [2] we clearly state that “The low intensity peaks at 100 and 68 ppm are attributed according to our XPS data to oxygenated carbon, mainly to lactone and epoxy groups, respectively”, and in addition highlight these peaks in Fig. 6 and list their parameters in Table 3 of Ref. [2]. Thus, such approach provides by definition that the NMR data are completely consistent with the XPS results.
Finally, Freitas demonstrates [1] the comparison of spectral simulations applying CSA MAS model to fit the experimental 13C MAS NMR spectrum of milled graphite measured at the MAS speed of 14 kHz that is smaller than in our work. The author aims to show that (1) “the interactions other than CSA (e.g., the 13C–13C dipolar coupling) play an important role in the formation of the spinning sidebands manifold in the spectra of [2]”, and (2) “the differences in the intensities of the first-order spinning sidebands are quite small for a large variation” in δani.
The much weaker intensity of ssb in Ref. [1] can be an indication of whatever, but definitely no argument for the presence of the interactions other than CSA in the spectra of [2]. The main reason is that the samples reported in our study [2] are fine grained graphitic materials consisting of the folded particles of various sizes, and therefore, cannot be directly compared to common graphites. On the other hand, the values of − 80 to − 120 ppm used by Freitas to fit the spectrum in [1] are far below the anisotropy value of − 163 ± 5 ppm reported in [9] that points out to the fact that milled graphite is not a reference system to compare the ssb intensities.
Our next critic point relates to the way of visual presentation of the spectral simulation in [1], where the author places fit and the spectrum one above each other to demonstrate “a subtle change in the intensity of the spinning sidebands” for a large variation in δani. However, our careful examination and overlaying of the experimental and simulated spectra reported in Ref. [1] showed a poor matching of both intensity and line width in the region of the central component and evidenced inappropriateness of such approach.
In conclusion, we demonstrated that criticism raised in Ref. [1] in respect to the fit model of 13C MAS NMR spectra of the 13C-enriched graphitic materials discussed in [2] is completely inappropriate. We showed that the experimental conditions used in [2] allow us to rule out the consideration of the 13C–13C homonuclear dipolar couplings in fitting the data. The presence of the spectral components beyond the typical graphitic range reported in Ref. [2] is not surprising and can be supported by the experimental and theoretical studies. While criticizing “such an enormous isotropic shift of the resonance lines from the usual shift range expected from sp2 carbons”, the author of [1] reported some time ago the 13C signals at 60 ppm for unmilled graphites [7]. Moreover, for ab initio calculations of NMR parameters in graphitic materials, the effects of the magnetic shielding originating from electronic current densities induced by the application of an external magnetic field must be taken into account. The observed in our spectra spinning sidebands originate from the CSA interactions, and therefore the CSA MAS model is most appropriate to fit the 13C spectra reported in [2]. Moreover, the CSA fit parameters deliver the important information for the interpretation of the results. Finally, simulation of the 13C MAS NMR spectrum in [1] is very confusing and misleads the readership into believing that the values reported in our work [2] are inaccurate and therefore meaningless. To sum up, none critical point in Ref. [1] is able to disprove the claims of our work reported in Ref. [2].
Change history
03 August 2021
A Correction to this paper has been published: https://doi.org/10.1007/s00723-021-01387-2
References
J.C.C. Freitas, Appl. Magn. Reson. 50, 1245 (2019)
Y.V. Fedoseeva, A.V. Okotrub, V.O. Koroteev, Y.M. Borzdov, Y.N. Palyanov, Y.V. Shubin, E.A. Maksimovskiy, A.A. Makarova, W. Münchgesang, L.G. Bulusheva, A. Vyalikh, Carbon 141, 323 (2019)
V.O. Koroteev, W. Münchgesang, Y.V. Shubin, Y.N. Palyanov, P.E. Plyusnin, D.A. Smirnov, K.A. Kovalenko, M. Bobnar, R. Gumeniuk, E. Brendler, D.C. Meyer, L.G. Bulusheva, A.V. Okotrub, A. Vyalikh, Carbon 124, 161 (2017)
A. Vyalikh, L.G. Bulusheva, G.N. Chekhova, D.V. Pinakov, A.V. Okotrub, U. Scheler, J. Phys. Chem. C 117, 7940 (2013)
A.V. Okotrub, M.A. Kanygin, V.O. Koroteev, S.G. Stolyarova, D.V. Gorodetskiy, Y.V. Fedoseeva, I.P. Asanov, L.G. Bulusheva, A. Vyalikh, Synth. Met. 248, 53 (2019)
A. Vyalikh, V.O. Koroteev, W. Münchgesang, T. Köhler, C. Röder, E. Brendler, A.V. Okotrub, L.G. Bulusheva, D.C. Meyer, ACS Appl. Mater. Interfaces 11, 9291 (2019)
M.A. Vieira, G.R. Gonçalves, D.F. Cipriano, M.A. Schettino Jr., E.A. Silva Filho, A.G. Cunha, F.G. Emmerich, J.C.C. Freitas, Carbon 98, 496 (2016)
M.M. Golzan, P.B. Lukins, D.R. McKenzie, A.M. Vassallo, J.V. Hanna, Chem. Phys. 193, 167 (1995)
C. Goze-Bac, S. Latil, P. Lauginie, V. Jourdain, J. Conard, L. Duclaux, A. Rubio, P. Bernier, Carbon 40, 1825 (2002)
M.A.L. Marques, M. d’Avezac, F. Mauri, Phys. Rev. B 73, 1693 (2006)
D. Sebastian, K.N. Kudin, ACS Nano 2, 661 (2008)
F. Simon, C. Kramberger, R. Pfeiffer, H. Kuzmany, V. Zólyomi, J. Kürti, P.M. Singer, H. Alloul, Phys. Rev. Lett. 95, 17401 (2005)
T.L. Makarova, Semiconductors 38, 615 (2004)
D.L. Vanderhart, W.L. Earl, A.N. Garroway, J. Magn. Reson. 44, 361 (1981)
D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.‐O. Durand, B. Bujoli, Zh. Gan, G. Hoatson, Magn. Reson. Chem. 40, 70 (2002)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised due to retrospective open access order.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vyalikh, A., Okotrub, A.V., Koroteev, V.O. et al. Comment on “On the Difficulties and Pitfalls with the Analysis of Solid‑State 13C NMR Spectra in Graphitic Materials”. Appl Magn Reson 52, 81–90 (2021). https://doi.org/10.1007/s00723-020-01215-z
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-020-01215-z