
Abstract
The various stages of the tetraethyl orthosilicate-based sol–gel process were investigated using electron paramagnetic resonance (EPR) spectroscopy with Cu(II) cations as a spin probe. The latter were introduced to the starting reaction mixture in the form of various copper(II) salts containing anions of different basicity (CuCl2, Cu(ac)2, or CuSO4). At the various defined stages of the sol–gel process, the experimental EPR spectra, recorded at both ambient and liquid nitrogen temperature, were found to be a superimposition of three main types of individual subspectra (Γ 1, Γ 2, and Φ), which reflect the different local environment in which the Cu(II) ions were located. The spin Hamiltonian parameters of each individual subspectrum remained identical, within experimental error, throughout the various stages of the sol–gel process. In contrast, the relative proportion of the individual subspectra varied significantly as the sol–gel process proceeded, from which the liquid-state to solid-state transition could be monitored as it occurred in the sol–gel reaction medium. Identical results were obtained, irrespective of the nature of the copper(II) salt employed. The results demonstrate that the EPR method provides an effective means with which to monitor the sol-to-gel transition from the viscous, colloid suspension to the final viscoelastic gel.
Similar content being viewed by others

1 Introduction
The sol–gel process is a promising technique for the synthesis of a large variety of advanced materials including high-purity silica glasses, which may be polycrystalline or composite in nature, for optics and optoelectronic applications. Since the sol–gel method operates at low temperature, a fine control of the chemical composition of these nanomaterials is possible [1].
The tetraethyl orthosilicate (TEOS)-based or tetramethyl orthosilicate (TMOS)-based sol–gel reaction mixture is electron paramagnetic resonance (EPR)-silent, since it is largely devoid of molecules containing unpaired electrons. Hence, to use EPR in the study of sol–gel conversions, it is necessary to introduce paramagnetic species, such as transition metal ions, into the sol–gel reaction mixture to serve as a spin probe of the medium. Those ions most commonly employed for this purpose are Cu(II), Mn(II), VO(II), and less frequently, Ti(III), Fe(III), Cr(III), Cr(V), Co(II), Ni(II) [2]. The corresponding EPR spectra are found to be very sensitive to changes in the local environment of these paramagnetic centres [3, 4], which may be thus monitored, for which purpose Cu(II) ions appear to be those most frequently used as a spin probe [5–38]. While it has been amply demonstrated that Cu(II) ions are very sensitive spin probes for the investigation of the structural changes that occur in silica xerogels during their calcination [5–14, 20–25, 29–31], they have been used far less frequently to investigate the earlier stages of the sol–gel process (initial reaction mixture → sol → gel conversion) [12, 14, 25].
Tominaga et al. [5] studied EPR spectra of Cu(II) ions that had been incorporated into silica gel via ion exchange technique, and recorded two different sets of EPR signals following water absorption, one of which was assigned to a distorted octahedral coordination geometry, and the other to a distorted tetrahedral configuration. Darab and McCrone [6, 9] analysed the multicomponent Cu(II) EPR spectra recorded both at room temperature and liquid nitrogen temperature, from which they concluded that a superposition of three main types of individual subspectra appeared in the various stages of the sol–gel process, according to the different environments in which the Cu(II) paramagnetic ions were contained. The authors concluded that the Cu(II) ions were not incorporated into the silica network, but instead remained in the pores of the TEOS-based xerogels, where they occupied surface sites [6, 9]. EPR results were reported for organically modified silica gels and xerogels, measured as a function of calcination temperature using Cu(II) as a spin probe, in a series of papers published by Klonkowski et al. [8, 10, 11, 16, 21, 22, 31]. In the majority of cases, the Cu(II) EPR spectra were observed to consist of the superposition of two or more individual subspectra, whose relative proportion varied. In a study by Shames et al. [12], it was found that the EPR spectra of Cu(II) ions and their o-phenanthroline complexes were insensitive to the changes that occur during the sol → gel → xerogel transition in a sol–gel reaction mixture of the following composition: tetramethoxysilane (TMOS), methanol, H2O, with NaOH present as a catalyst. In contrast, we reported our preliminary results on the successful monitoring of the sol → gel → xerogel transition in the sol–gel reaction mixture (tetraethoxysilane (TEOS), ethanol, H2O, with HCl present as a catalyst) using EPR spectroscopy with Cu(II) as a spin probe, at the 19th International Conference on Coordination Chemistry, Smolenice, Slovakia [39]. The present full paper is a consolidation of our work on this topic.
The sol–gel process may be thought to occur via a sequence of steps, of which the following are dominant: (1) nucleation and growth of the sol particles; (2) coagulation of the sol particles to form larger aggregates; (3) gel network formation in the sol–gel system [2]. The overall sol–gel transformation is characterised by dramatic increases in the bulk viscosity of the reaction mixture [24]. Since the paramagnetic spin-probe molecules [e.g. hydrated Cu(II), Mn(II), or VO(II) ions] remain free to reorient in the solvent phase, between the growing and agglomerating sol particles, typical “liquid-type” EPR spectra are recorded at ambient (room) temperatures (RT). As the process advances, the transition metal ion complexes may become fixed to the surface of the growing gel particles and/or localised in the solvent liquid which fills the pores of the developing gel network. Those ions that are attached to larger particles are immobilised on the EPR timescale, and give EPR spectra with a typical “solid-state” appearance, even at RT [20, 24, 25]. The mechanisms by which such attachment might occur are discussed in an excellent review paper, by Dyrek and Che [40]. Using EPR spectroscopy, Martini et al. [41–43] studied the mobility of [Cu(H2O)6]2+ ions, in hydrated silica gels with varying pore diameters in the range of 4–100 nm, and found that the spin-probe mobility depended on the pore radii in all wet gel, dry gel and xerogel samples. They concluded that the adsorbed water behaves similarly to bulk water in environments where the pore diameters are greater than 10 nm, while in smaller pores, its mobility is more restricted [43]. This may be explained in terms of a greater degree of immobilisation of the water layers that are close to the pore surface, as compared with water contained more deeply within the pore. On freezing, two different types of water could be identified: (1) “unfreezable water”, which is confined to the first ten layers of water molecules, in which the normal (bulk) ice structure cannot be formed because of the effect of the surface interaction on the water structure; (2) “freezable water”, which is bulk water contained in pores larger than 4 nm, which does undergo crystallisation, although with a depressed freezing point as a result of long-range meniscus effects [43].
Our previous EPR work on silica xerogels was focussed primarily on investigating the calcination process as a function of the firing temperature, along with the effect of subsequent ageing on the material, using Cu(II) as a spin probe [13, 14, 30]. In the present study, we have monitored the initial stages of the sol–gel process, with particular emphasis on the transformation of the starting reaction mixture from the liquid state to the solid state, and with due consideration being paid to the possible location of hydrated complexes [Cu(H2O)6]2+ in the wet and dry gels. Likewise, we compare three different sol–gel sample sets, in which Cu(II) ions from various copper(II) salts with anions of different basicity (CuCl2, Cu(ac)2, or CuSO4) were employed as the spin probe.
2 Experimental
The samples were prepared by the same procedure and subsequently the EPR measurements were performed as described in details in our previous paper [44]. A very brief summary: in the present work, the Cu(II) spin probe in the form of the three various copper(II) salts, CuCl2 2H2O (Sigma-Aldrich, 98.0 %), Cu(ac)2 H2O (Sigma-Aldrich, 98.0 %) or CuSO4 5H2O (Sigma-Aldrich, 98.0 %), was incorporated into the binary mixture of TEOS (Sigma-Aldrich, 98.0 %) and ethanol for UV (MicroChem) before hydrolysis began, so to give a final Cu(II) concentration of ca 1 mmol. Then, the re-distilled water containing HCl (MicroChem) was added dropwise to catalyse the hydrolysis. Thus, a sol with a yellow-green or green-blue colour was obtained, depending on which copper(II) salt was used.
The hydrolysis and condensation reactions (in air at RT) proceed in a closed glass containers with a plastic cover with holes in it. Under these conditions, the gelation was completed after about 2 weeks giving “wet gel”, which was dried (in a furnace at 100 °C in air for 12 h) giving “dry gel”. In the defined time (immediately, then hourly, up to 12 h, and the daily, up to 30 days), the sample sets were prepared for EPR experiment. Likewise, the “dry gel” was hydrated by storing them for 3 days in a container with an atmosphere saturated with water vapour at RT. In addition, after hydrolysis began in the batch reactor, a small part of the reaction mixture was taken and placed into a quartz EPR capillary, which was then closed.
The EPR spectra were recorded on a Bruker X-band EPR spectrometer at both room temperature (RT) and at liquid nitrogen temperature (77 K) [44, 45]. A special procedure was used to precisely position the quartz EPR sample tubes within the microwave cavity, as was described previously [46, 47]. All EPR spectra were processed using WinEPR [48] and simulated by SimFonia [49] software. The spin Hamiltonian parameter values, which were obtained from the experimental Cu(II) EPR spectra, were then further refined by computer simulation [49, 50]. The multicomponent EPR spectra were analysed as a linear combination of individual subspectra [50]. The control samples prepared without Cu(II) ions were EPR silent. No differences were found between the results obtained using different copper(II) salts containing anions of different basicity (CuCl2, Cu(ac)2, or CuSO4) in the starting reaction mixture. Accordingly, the Cu(II) EPR spectra obtained in the TEOS-based sol–gel reaction mixture doped with Cu(II) ions in the form of CuCl2 presented, below, may be taken as being representative of the results overall.
3 Interpretation and Simulation of Multicomponent Cu(II) EPR Spectra
Following Darab and McCrone [6, 9], the multicomponent experimental Cu(II) EPR spectra of the sol–gel process can be interpreted in terms of three main forms of individual subspectra, which correspond to different environments of the paramagnetic Cu(II) ions. These are, a superposition of two types of axially symmetric subspectra, (1) Γ 1-type, which reflects a highly condensed and rigid polymer network based on the Si–O–Si skeleton, and (2) Γ 2-type, in which the gel skeleton has a looser structure, and contains many pores of differing size, which can be open to or isolated from the external environment, and through which the Cu(II) ion can easily move. There is also (3) a broad singlet present, Φ-type, which is from Cu(II) ions that are located in liquid-state environments of varying viscosity.
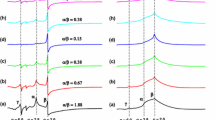
In Fig. 1, we presented the series of computed two-component EPR spectra of Cu(II) ions containing varying relative proportions of the simulated individual EPR subspectra: (a) Γ 1 and Γ 2; (b) Γ 2 and Φ, in the range 0.1–0.9. It may be seen that when the relative proportion of the Φ subspectrum increases to 0.8, the resolution of the hyperfine splitting in the Γ 2 subspectrum is lost and only the broad “pseudosinglet” is apparent. The spin Hamiltonian parameters of individual subspectra used in the simulations were taken form our previous works [13, 14, 30] and are summarised in the Table 1.

Computed two-component Cu(II) EPR spectra calculated for the various relative contributions (0.1–0.9) of the simulated individual EPR subspectra a Γ 1 and Γ 2, b Γ 2 and Φ
4 Results and Discussion
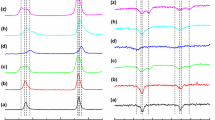
The experimental EPR spectra measured at (a) room temperature (RT) and (b) liquid nitrogen temperature (77 K), as were recorded at the various stages of the sol–gel process, are illustrated in Fig. 2. The details of the parallel hyperfine lines (m I = −3/2, −1/2, 1/2, 3/2) of the multicomponent EPR spectra for the various stages (a–d) of the sol–gel process, hydrated dry gel (h) and sol–gel formed in the sealed capillary (z), with an indication of the Γ 1, Γ 2(Γ 2′, Γ 2′′), and Φ lines are shown in Fig. 3.

Experimental Cu(II) EPR spectra recorded at a RT and, b 77 K for the various stages of the sol–gel process a immediately after hydrolysis started (reaction mixture, solution of low viscosity), b sol (colloidal suspension, solution of high viscosity), c wet gel (viscoelastic solid), d dry gel (powder solid state), h hydrated dry gel and z process occurring in sealed capillary

Expansion of the parallel hyperfine lines (m I = −3/2, −1/2, 1/2, 3/2) of the low field part of the 77 K Cu(II) EPR spectra (shown in Fig. 2b) with the indication of the individual subspectra, Γ 1, Γ 2(Γ 2′, Γ 2′′) and Φ
As recorded at RT (Fig. 2a), in the water-like reaction mixture of low viscosity, stage (a), and in the highly viscous, colloidal suspension, stage (b), the hyperfine structure of the Cu(II) resonance lines (quartet) from 63,65Cu (I = 3/2), was not resolved, and only the asymmetric wide “pseudosinglet” (g ef = 2.146 ± 0.002, \(\Delta\) B pp = 15.05 ± 0.02 mT) was observed. This is most likely due to a combination of the electron–electron exchange interaction between different Cu(II) ions, and the dynamic Jahn–Teller effect [51, 52]. Other than a marginal increase in the signal intensity and peak-to-peak line width (\(\Delta\) B pp = 15.59 ± 0.02 mT) measured at stage (b), the spectrum was essentially identical with that recorded at stage (a). On the basis of line shape simulations (see Fig. 1b), it was determined that this asymmetric “pseudosinglet” is a combination of two individual subspectra, dominantly of Φ-type, with a relative minor contribution from the Γ 2-type. However, once the sol-to-gel transition was complete, i.e. wet gel (c), significant changes were observed in the EPR spectra of the Cu(II) spin probe, as a result of a decrease in the relative proportion of the Φ subspectrum, while the proportion of the Γ 2 subspectrum increased significantly. The effect of drying can be seen from the EPR spectra of the wet gel (c) and dry gel (d), during which the relative contribution of the Φ subspectrum decreased further and indeed to the limit of resolution. Thus, in the dry gel (d), the EPR spectrum consists of the superposition of two axially symmetric subspectra (Γ 1 and Γ 2) and a broad “pseudosinglet” of low intensity from the Φ subspectrum. We note that these EPR spectra are very similar to those recorded previously from Cu(II) ions in dry gels and xerogels at RT [5–14, 24, 25, 30]. The significant differences amongst the spectral features clearly demonstrate the viscosity and structural changes that occur during the sol-to-gel transition, stages (b–c), and during the wet gel drying, stage (d). The EPR spectra recorded from the control sample, (z), in which the sol–gel process was allowed to proceed inside a closed capillary, shows a very similar spectral shape to that obtained in the wet gel (c), i.e. with a poorly resolved hyperfine splitting, and a marked increase in the relative contribution from the Φ subspectrum. Equivalently, the dry gel hydration procedure, sample (h), resulted in a line shape that was very similar to that recorded from the wet gel (c), i.e. with an appreciable increase of relative contribution from the Φ line.
As measured at 77 K (Fig. 2b), a parallel hyperfine splitting is fully or partly resolved in the EPR spectra measured for samples taken at all stages (a–d) of the sol–gel process, for the hydrated gel (h) and for the control (z) sample. In Fig. 3, the details of the parallel hyperfine lines (m I = −3/2, −1/2, 1/2, 3/2) of the multicomponent EPR spectra clearly show the presence of two new axially symmetric subspectra of the Γ 2-type {Γ 2′ (g⊥ = 2.073 ± 0.002, g|| = 2.352 ± 0.002, A|| = 13.17 ± 0.02 mT) and Γ 2′′ (g⊥ = 2.075 ± 0.002, g|| = 2.409 ± 0.002, A|| = 11.82 ± 0.02 mT)}, which were not detected at RT. Due to the low intensity of some of the Γ 1, Γ 2(Γ 2′, Γ 2′′) EPR lines, and their partial overlap with one another, it was not possible to make a precise determination of their relative proportions. It is possible that the Γ 2(Γ 2′, Γ 2′′) signals arise from Cu(II) ions that are situated within pores of varying dimension, and these can be open to, or isolated from, the external environment. Other than the fact that the total intensity of the EPR signal increased marginally, as did the relative proportion of the Γ 2(Γ 2′, Γ 2′′) resonances, there was no substantial difference between the spectra recorded from samples taken at stage (a) or stage (b). Once the sol-to-gel conversion was complete, wet gel (c), the intensity of the Γ 2(Γ 2′, Γ 2′′) subspectra was found to decrease, while that of the Γ 1 subspectrum increased. In the dry gel (d), while the signal from the Γ 1 subspectrum is dominant, minor Γ 2(Γ 2′, Γ 2′′) resonances are also apparent. The EPR spectra recorded at 77 K from the sealed capillary (z) were practically identical with those from the wet gel, stage (c), in consequence of the presence of solvents (ethanol and water) which remain within the gel pores as the sol–gel process advances inside the closed capillary. The EPR spectra recorded from the sample of hydrated dry gel (h) were similar to those observed from the wet gel (c) and the sealed capillary (z).
To assess the effect of hydration and dehydration on the material, the wet gel (c) was dried in air at 100 °C to form the dry gel (d), which was then hydrated for 3 days at RT by placing samples of it in a container with an atmosphere saturated with water vapour (h), so that water molecules may be imbibed into the gel pores. The EPR spectra show that once the solvent has been evaporated from the wet gel samples at 100 °C, the water molecules are removed from the hydration sphere of the Cu(II) ions, which become fully immobilised on the pore surface. When the dry gel is hydrated, water again enters the pores to re-create [Cu(H2O)5]2+ complexes, which interact with silanol groups on the pore surface. This process of hydration and dehydration is completely reversible, since the form of the EPR spectra, which was changed by drying the wet gel (c) in air at 100 °C, gradually reappeared in the dry gel (d) after a few days in the presence of an atmosphere saturated with water vapour at RT. No difference in the spectral parameters were found between a sample of the dry gel that had been hydrated for 3 days, and recorded after 3 days, or for a dry gel sample that had been hydrated over a period of 7 days and then measured. In accord with the literature data [6, 9, 12, 24, 25, 29, 53–55], the hydrated copper complexes, [Cu(H2O)6]2+ are not strongly linked to the polymer surface, remain solvated and are free in tumbling motion in the gel pores. However, the effective viscosity of the liquid phase localised in these pores is higher than the viscosity of the earlier reaction mixture [55–58].
In the TEOS-based sol–gel reaction mixture, Cu(II) ions coordinate with solvent molecules to form hexa-coordinate complexes, which are usually free to tumble within the liquid medium. This mobility is strongly impeded when they are in the vicinity of a solid surface or inside narrow gel pores [40, 54]. Bassetti et al. [43] employed EPR spectroscopy to study the mobility of water molecules adsorbed in differently sized silica gel pores (within the range 4–100 nm) containing Cu(II) ions as a spin probe. Independent of the pore size, isotropic EPR spectra of Cu(II) ions were recorded at RT, which consisted of a broad singlet centred at g ≈ 2.18 (no hyperfine splitting was resolved), which is very similar to the spectra typically recorded from dilute aqueous solutions of Cu(II) salts, and can be attributed to the [Cu(H2O)5]2+ complexes present in silica gel cavities that are filled by water. Since we have similarly observed isotropic EPR spectra of Cu(II) ions as a broad “pseudosinglet” in the reaction mixture (a) and the sol (b) at RT, our results are consistent with these observations. However, the EPR spectra recorded at RT in the wet gel (c), hydrated dry gel (h) and sealed capillary (z) show traces of an axially symmetric EPR signal, the proportion of which is significantly increased in the dry gel sample (d). Since these spectra are of typical solid-state appearance, they are probably from Cu(II) ions that are immobilised in gel pores with diameters of less than 6 nm [43]. That this signal persists at RT indicates that some portion of the Cu(II) ions is located in sites where the surface field is sufficiently high to immobilise ions even at ambient thermal energies.
The EPR spectra measured at RT from the (c, d, h, z) samples reveal the presence simultaneously of both liquid-state and solid-state features. We may deduce, therefore, that the mobility of the Cu(II) ions is reduced and some of them are immobilised, presumably those in the narrowest pores, giving rise to a solid-state type spectrum. In contrast, those ions that are in the largest pores, and hence at some distance from the surface, behave as though they are in a bulk liquid environment. In principle, the liquid-type spectrum should be converted to a solid-type spectrum when the correlation times for the tumbling are increased to the order of τ r = 10−8–10−9 s [43]. We note that EPR spectroscopy is sensitive to viscosities at the local molecular level, and reflects the space between the sol particles or the differing dimensions of the gel pores. Such a “microscopic” measure of the environment of Cu(II) ions is naturally rather different from the bulk “macroscopic” scale that is measured by a viscosity meter [20].
At 77 K, typical “frozen solution-type” and “frozen powder-type” Cu(II) EPR spectra were recorded from the samples (a, b), and (c, d, h, z), respectively, all of which can be interpreted in terms of the superimposition of three main individual subspectra, the axially symmetric Γ 1, Γ 2(Γ 2′, Γ 2′′) and broad singlet Φ, with their different relative contributions. The small differences observed in the spin Hamiltonian parameters of the Γ-type subspectra show that the [Cu(H2O)5]2+ complexes responsible for the axially resolved spectra are weakly, yet significantly, influenced by their interaction with the pore surface. The parameter values of the individual subspectra are very similar to those recorded for [Cu(H2O)5]2+ complexes in silica gels at 77 K, as reported in the literature [40–43, 54]. The present results confirm the view [40–43] that two different types of water are present in the silica gel systems at 77 K: crystalline water and amorphous water. Several regions have been identified depending upon the strength of the electric field produced by the walls of pores: (1) a region in which water is strongly immobilised even at room temperature (this region comprises the first three layers of water from the surface); (2) a more extended region with an increased water viscosity (involving about ten layers of water molecules); (3) a bulk water region where the water molecules are sufficiently distant from the surface (>2 nm) to avoid any reduction in their mobility, so that crystallisation occurs in the usual manner.
5 Conclusions
No differences were found between the results obtained using different copper(II) salts containing anions of different basicity (CuCl2, Cu(ac)2, or CuSO4) in the starting reaction mixture.
In the various stages of the sol–gel process, the experimental multicomponent EPR spectra of Cu(II) ions recorded both at room temperature and liquid nitrogen temperature were a superimposition of three main types of individual subspectra (Γ 1, Γ 2 and Φ), which reflect the different local environment of the Cu(II) ions. The spin Hamiltonian parameters of each individual subspectrum were, within experimental error, identical throughout the various stages of the sol–gel process. However, the relative contribution of these individual subspectra varied significantly, stage by stage, as the sol–gel process proceeded, which permitted us to monitor the changes that occurred in the sol–gel reaction mixture as it transformed from the liquid state to the solid state.
In accord with results previously reported in the literature [5–12, 24, 25] and from our own work [13, 14, 30], we may conclude that EPR spectroscopy using Cu(II) as a spin probe provides an effective method with which to monitor the evolution of the TEOS-based sol–gel process, including the sol-to-gel transition from the viscous, colloid suspension to the viscoelastic gel. We conclude that during the sol–gel process, the hydrated Cu(II) ions are contained in a solution-like environment where they can undergo a relatively free tumbling motion.
References
D. Trandafir, M. Vasilescu, S. Simon, J. Sol-Gel Sci. Technol. 63, 425–434 (2012)
C.J. Brinker, G.W. Scherer, Sol-Gel Science (Academic Press, New York, 1990)
J. Wong, C. Angel, Glass structure by spectroscopy (Marcel Dekker, Basel, 1976)
D.L. Griscom, Glass Sci. Technol., vol. 4B. (Academic Press, Boston, 1990)
H. Tominaga, Y. Ono, T. Keii, J. Catal. 40, 197–202 (1975)
J.G. Darab, R.K. MacCrone, J. Non Cryst. Solids 95&96, 1203–1210 (1987)
S. Ikoma, S. Tanako, E. Nomoto, H. Yokoi, J. Non Cryst. Solids 113, 130–136 (1989)
A.M. Klonkowski, C.W. Schlaepfer, J. Non Cryst. Solids 129, 101–108 (1990)
J.G. Darab, R.K. MacCrone, Phys. Chem. Glasses 32, 91–101 (1991)
A.M. Klonkowski, C.W. Schlaepfer, J. Non Cryst. Solids 149, 189–195 (1992)
A.M. Klonkowski, K. Koehler, C.W. Schlaepfer, J. Mater. Chem. 3, 105 (1993)
A. Shames, O. Lev, B. Iosofzon, J. Non Cryst. Solids 163, 105–114 (1993)
M. Liška, M. Mazur, H. Hulínová, P. Pelikán, M. Valko, I. Nerad, Ceram-Silicate 39, 69–72 (1995)
M. Mazur, M. Valko, R. Klement, P. Pelikán, Prog. Coord. Organometal. Chem. 3 (STU Press, Bratislava, 1997), pp 285–290
G. Córdoba, M. Viniegra, J.L. Fierro, J. Padilla, R. Arroyo, J. Solid State Chem. 138, 1–6 (1998)
A.M. Klonkowski, B. Grobelna, T. Widernik, A. Jankowska-Frydel, W. Mozgawapar, Langmuir 15, 5814–5819 (1999)
J.F.P. Robles, F.J.G. Rodrigues, J.M.Y. Limon, F.J.E. Beltran, Y.V. Vorobiev, J.G. Hernandez, J. Phys. Chem. Solids 60, 1729–1736 (1999)
V.B. Prokopenko, V.S. Gurin, A.A. Alexenko, V.S. Kulikauskas, D.L. Kovalenko, J. Phys. D Appl. Phys. 33, 3152–3155 (2000)
J.M. Yánez-Limón, J.F. Pérez-Robles, J. González-Hernández, Y.V. Vorobiev, J.A. Romano, F.C.G. Gandra, B.C. Da Silva, J. Sol-Gel Sci. Technol. 18, 207–217 (2000)
T. Breyer, F.W. Breitbarth, W. Vogelsberger, Bull. Polish Acad. Sci. Chem. 48, 325–336 (2000)
W.K. Jozwiak, E. Szubiakiewicz, A.M. Klonkowski, T. Widernik, W. Ignaczak, T. Paryjczak, Stud. Surf. Sci. Catal. 130, 3315–3320 (2000)
A.M. Klonkowski, T. Widernik, B. Grobelna, W.K. Jóźwiak, H. Proga, E. Szubiakiewicz, J. Sol-Gel Sci. Technol. 20, 161–180 (2001)
P.I. Paulose, J. Gin, T. Vinoy, J. Gijo, N.V. Unnikrishanan, M.K.R. Warrier, Bull. Mater. Sci. 25, 69–74 (2002)
W. Vogelsberger, A. Seidel, T. Brayer, Langmuir 18, 3027–3033 (2002)
T. Breyer, F.W. Breitbarth, W. Vogelsberger, J. Colloid Interf. Sci. 266, 153–159 (2003)
Z. Wang, Q. Liu, J. Yu, T. Wu, G. Wang, Appl. Catal. A Gener. 239, 87–94 (2003)
S. Mukherjee, A.K Pal. J. Non Cryst. Solids 341, 170–177 (2004)
O.B. Miled, C. Boissiere, C. Sanchez, J. Livage, J. Phys. Chem. Solids 67, 1775–1780 (2006)
L.D. Bogomolova, T.K. Pavlushkina, I.V. Morozova, Glass Ceram. 63, 254–258 (2006)
M. Mazur, J. Moncol, M. Kleinová, P. Stachová, M. Valko, Phys. Chem. Glasses 47, 278–282 (2006)
K. Kledzik, M. Jamrógiewicz, M. Gwiazda, E. Wagner-Wysiecka, J. Jezierska, J.F. Biernat, A.M. Klonkowski, Mater. Sci. Poland 25, 1041–1051 (2007)
D.H. Ji, X.J. Zou, D.F. Zheng, L.X. Wang, S.Y. Liu, W.C. Zhu, G.J. Wang, Z.L. Wang, Polish J. Chem. 82, 1097–1104 (2008)
J.H. Chen, F. Wang, N.S. Cheng, Z.J. Ding, Z.H. Fu, D.H. Yin, J. Inorg. Mater. 24, 695–701 (2009)
Q. Zhang, X. Liu, Y. Qiao, B. Qian, G. Dong, J. Ruan, Q. Zhou, J. Qiu, D. Chen, Opt. Mater. 32, 427–431 (2010)
F. Vignali, G. Predieri, E. Feci, S. Palanti, M.C. Baratto, R. Basosi, E. Callone, K. Müller, J. Sol-Gel Sci. Technol. 60, 445–456 (2011)
A. Dehelean, S. Rada, V. Danciu, E. Culea, M. Stan, A. Popa, O. Raita, AIP Conf. Proc. 1425, 65–68 (2012)
M.P. Pachamuthu, A. Ramanathan, K. Santhi, R. Maheswari, ACS Symp. Ser. 1132, 195–211 (2013)
G. Sivasubramanian, C. Shanmugam, V.R. Parameswaran, J. Porous Mater. 20, 417–430 (2013)
J. Mazur, M. Švorec, M. Kleinová, Valko. Prog. Coord. Bioinorg. Chem. 6, 317–322 (2003)
K. Dyrek, M. Che, Chem. Rev. 97, 305–331 (1997)
G. Martini, V. Bassetti, J. Phys. Chem. 89, 2505–2511 (1979)
G. Martini, V. Bassetti, J. Phys. Chem. 89, 2511–2515 (1979)
V. Bassetti, L. Burlamacchi, G. Martini, J. Phys. Chem. 101, 5471–5477 (1979)
M. Mazur, M. Kleinová, J. Moncol, P. Stachová, M. Valko, J. Telser, J. Non Cryst. Solids 352, 3158–3165 (2006)
M. Mazur, L. Husáriková, C.J. Rhodes, M. Valko, J. Sol-Gel Sci. Technol. 76, 110–119 (2015)
M. Mazur, M. Valko, R. Klement, H. Morris, Anal. Chim. Acta 333, 249–252 (1996)
M. Mazur, M. Valko, H. Morris, R. Klement, Anal. Chim. Acta 333, 253–265 (1996)
H. Thiele, J. Etstling, P. Such, P. Hoefer, WINEPR (Bruker Analytic Gmb, Rheinstetten, 1992)
R.T. Weber, WINEPR SimFonia (EPR Division, Bruker Instr. Inc., Billerica, 1995)
P. Pelikán, M. Liška, M. Valko, M. Mazur, J. Magnet. Reson. 122, 9–15 (1996)
H.A. Jahn, E. Teller, Proc. R. Soc. 161, 220–235 (1937)
L.B. Bersuker, Coord. Chem. Rev. 14, 357–412 (1975)
A. Shames, O. Lev, B. Iosefzon-Kuyavskaya, J. Non Cryst. Solids 175, 14–20 (1994)
K. Dyrek, A. Adamski, Z. Sojka, Spectrochem. Acta Part A 54, 2337–2348 (1998)
L.D. Bogomolova, A.V. Jackin, N.A. Krasilnikova, J. Non Cryst. Solids 241, 13 (1998)
N. Abiddi, B. Deroide, J.V. Zanchetta, D. Bourret, H. Elmkami, P. Rumori, Phys. Chem. Glasses 37, 149 (1996)
N. Abiddi, B. Deroide, J.V. Zanchetta, Nucleonika 42, 505 (1997)
P. Rumori, B. Deroide, N. Abiddi, H. Elmkami, J.V. Zanchetta, J. Phys. Chem. Solids 59, 959 (1998)
Acknowledgments
This work was supported by the Scientific Grant Agency of the Slovak Republic (Projects VEGA 1/0765/14 and 1/0041/15).
Author information
Authors and Affiliations
Corresponding author
Additional information
Dedicated to the 70th anniversary of the discovery of Electron Paramagnetic Resonance by Professor E. K. Zavoiskij.
Rights and permissions
About this article

Cite this article
Mazúr, M., Husáriková, L., Valko, M. et al. Monitoring the Tetraethyl Orthosilicate (TEOS)-Based Sol–Gel Process with Cu(II) Ions as a Spin Probe. Appl Magn Reson 47, 1–12 (2016). https://doi.org/10.1007/s00723-015-0724-x
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-015-0724-x