
Abstract
In the course of an exploratory study on the quaternary system Na2O-K2O-CaO-SiO2 single crystals of the first anhydrous sodium potassium calcium silicate have been obtained from slow cooling of a melt in the range between 1250 and 1050 °C. Electron probe micro analysis suggested the following idealized molar ratios of the oxides for the novel compound: K2O:Na2O:CaO:SiO2 = 1:1:12:8 (or KNaCa6Si4O15). Single-crystal diffraction measurements on a crystal with chemical composition K1.08Na0.92Ca6Si4O15 resulted in the following basic crystallographic data: monoclinic symmetry, space group P 21/c, a = 8.9618(9) Å, b = 7.3594(6) Å, c = 11.2453(11) Å, β= 107.54(1)°, V = 707.2(1) Å3, Z = 2. Structure solution was performed using direct methods. The final least-squares refinement converged at a residual of R(|F|) = 0.0346 for 1288 independent reflections and 125 parameters. From a structural point of view, K1.08Na0.92Ca6Si4O15 belongs to the group of mixed-anion silicates containing [Si2O7]- and [SiO4]-units in the ratio 1:2. The mono- and divalent cations occupy a total of four crystallographically independent positions located in voids between the tetrahedra. Three of these sites are exclusively occupied by calcium. The fourth site is occupied by 54(1)% K and 46%(1) Na, respectively. Alternatively, the structure can be described as a heteropolyhedral framework based on corner-sharing silicate tetrahedra and [CaO6]-octahedra. The network can build up from kröhnkite-like [Ca(SiO4)2O2]-chains running along [001]. A detailed comparison with other A2B6Si4O15-compounds including topological and group-theoretical aspects is presented.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The ternary systems Na2O-CaO-SiO2 and K2O-CaO-SiO2 have been studied for more than 90 years. Especially the sodium containing system has been in the focus of many research activities because of its fundamental importance for the production of commercial soda-lime-glasses (Varshneya 1994; Shelby 2009), which are the classical mass-produced types for windows and containers. However, the crystalline counterparts have also found quite a number of applications in Materials science. Na2CaSiO4 or Na2Ca2Si3O9 (combeite), for example, have been reported as major phases in bioactive glass ceramics or as host materials for inorganic phosphors, whereas overlapping fans of needles of Na2Ca3Si6O16 (devitrite) crystals have been successfully developed for manufacturing of low-cost optical diffusers (Kaygili 2014; Fujikura et al. 2012; Liu et al. 2014; Butt et al. 2014). The corresponding potassium system, on the other hand, has seen a revival of interest only recently. This is primarily related to the constantly increasing usage of renewable fuels such as biomass for the production of heat and/or electricity. Actually, the variation in composition of the silicate fraction of the bottom ashes and slags occurring during biomass combustion can be conveniently described in the triangle diagram K2O(+ Na2O)-CaO(+ MgO)-SiO2. Consequently, the corresponding simplified ternary phase diagram has been frequently used for the interpretation of (i) melt formation/composition and (ii) the phase content at sub-solidus conditions (Lindberg et al. 2013 and references cited therein). Both aspects have implications for process control as well since slagging, fouling or sintering can cause severe problems in practical operation (deposition on or corrosion of heat transfer surfaces as well as bed agglomeration in fluidized beds) (Lindberg et al. 2013).
To the best of our knowledge, for the more complex quaternary system Na2O-K2O-CaO-SiO2 no thermodynamic or phase-analytical data are available. This is somehow surprising because - at least among the amorphous materials of this system - it is known that the simultaneous presence of two different monovalent cations Na+ and K+ in the melt and the resulting glasses can induce dramatic changes in certain physicochemical properties (Dilmore et al. 1978; Grund and Jonson 2009). Actually, the term mixed alkali effect was coined a long time ago in silicate glass research to describe that chemical durability, electric conductivity or viscosity can vary in an extremely non-linear way if one alkali oxide is gradually substituted by another one (Shelby 2009). Furthermore, soda-potash-lime silicate glasses play also a role in archeology: the earliest known ancient Chinese glasses belong to this group of materials (Gan et al. 2006).
For the crystalline phases the situation is less understood. So far, not a single sodium-potassium-calcium silicate has been reported. However, simple structural extrapolations from the limiting calcium containing ternary systems have to be treated with care. First of all, the number of real ternary phases in the sub-systems Na2O-CaO-SiO2 and K2O-CaO-SiO2 is large (Kahlenberg and Maier 2016; Schmidmair et al. 2015) and for some phases reported in the older literature a final proof of existence is still missing. Furthermore, many of the well characterized compounds in the system Na2O-CaO-SiO2 have no direct chemical counterparts in the potassium system. Even for those cases where identical stoichiometries have been observed potential structural relationships between the two endmembers cannot be predicted. K2Ca2Si2O7 and Na2Ca2Si2O7, for example, represent different structure types based on [Si2O7]-groups (Arroyabe and Kahlenberg 2011) and 1:1 mixtures of [Si3O10]- and [SiO4]-moieties (Kahlenberg and Hösch 2002), respectively. K2Ca6Si4O15 (Arroyabe et al. 2009a) and Na2Ca6Si4O15 (Kahlenberg and Maier 2016) have related heteropolyhedral frameworks. Nevertheless, the topology of the networks is different. On the other hand, Na4CaSi3O9 (Fischer and Tillmanns 1984) and K4CaSi3O9 (Arroyabe et al. 2009b) are isotypic. However, the formation of a potential solid-solution series has not been studied.
In summary one can say that the phase equilibria and the structural landscape of the system Na2O-K2O-CaO-SiO2 is rather uncharted territory. The main aim of the present work was to make a first step to fill this gap in solid-state knowledge. This paper reports the synthesis and the results of the structural characterization of the first quaternary phase of the Na2O-K2O-CaO-SiO2 system.
Experimental details
Synthesis experiments for a sample with nominal composition (Na0.25K0.75)2Ca2Si2O7 were based on the following fine chemicals: Na2CO3 (Merck, 99.9%), CaCO3 (Merck, >99.9%), K2CO3 (Alfa Aesar, 99.997%) and SiO2 (AlfaAesar, 99.995%). Before weighing on an analytical balance (Mettler Toledo LabStyle 204), the educts were dried at 300 °C for 12 h. A total of 3 g of the stoichiometric mixture was homogenized for 15 min in a planetary ball mill (Fritsch Pulverisette 7) under ethanol. Subsequently, the sample was dried again at 300 °C for complete removal of the alcohol and, finally, stored in a desiccator.
In order to obtain some basic information of the high temperature behavior of the sample, 35.44 mg were studied in an open platinum crucible by thermogravimetric (TGA) and differential thermal (DTA) analysis. The experiment was performed in a Setaram SetsysEvolution 2400 apparatus with a resolution of the electromagnetic compensated torsion balance of 0,03 µg and controlled carrier as well as auxiliary gas flows (mixing ranges: 50/50 to 1/99). The TGA - DTA is coupled with a mass spectrometer (OmniStar QMS200, Pfeifer Vacuum Quadrupol). The sample holder is equipped with Pt/Pt90-Rh10 thermocouples. The temperature program contained a heating/cooling ramp of 5 °C per minute from room temperature to Tmax = 1300 °C and back to ambient conditions. The experiment was performed in air. The results can be summarized as follows: upon heating, the low–high quartz transformation (at 573 °C) as well as the stepwise CO2-release from the decomposition of the different carbonates in combination with starting solid-state reactions can be clearly identified (two effects with peak maxima at 748 and 1001 °C, respectively). A large endothermal signal (onset temperature: 1225 °C) indicated the formation of melt. At the maximum temperature of the experiment melting was still in progress. During cooling only one sharp intense exothermic effect could be detected (onset: 1125 °C, peak maximum: 1123 °C).
Single-crystal growth experiments were initiated in a resistance-heated chamber furnace using the following set of parameters: 1 g of the educt mixture confined in a lid-covered 50 ml platinum crucible was fired from 25 °C with a heating ramp of 50 °C/h to 1250 °C and annealed for 2 h. Subsequently, the sample was cooled with 5 °C/h to 1050 °C and immediately quenched in air by removing the crucible from the furnace. Weight loss was determined from weight differences before and after heating. The comparison of the theoretical (assuming a complete disintegration of the carbonates) and the experimental values revealed a pronounced difference: 29.31 wt% vs. 37.06 wt%. This discrepancy of about 7.7 wt% has to be attributed to additional K2O/Na2O-losses which are known to be the most volatile oxide components of this system. Notably, the corresponding difference in the DTA was much less distinct (2.1 wt%) although the maximum temperature was about 50 °C higher. This observation can be explained by the considerable shorter total run time of the heating/cooling cycle during thermal analysis.
A first optical examination of the shape of the quenched sample in the crucible indicated the presence of a melt phase in the course of the high-temperature treatment. After mechanical removal of the solidified melt cake from the container, it was slightly crushed in an agate mortar and further checked under a polarizing binocular. The visual inspection pointed to the presence of at least two different phases: (i) birefringent crystals up to 100 µm in diameter and (ii) an isotropic glassy matrix.
A part of the material from the growth experiment was further analyzed using X-ray powder diffraction (see Fig. 1). Data were collected on a Bruker D8-Discover diffractometer using a Cu-tube operated at 40 kV and 40 mA, a primary beam Ge-monochromator and a LYNXEYE™ silicon strip detector. Data acquisition was performed in Bragg–Brentano θ-2θ geometry at ambient temperature in the range between 3 and 135 °2θ with a counting time of 2 s per step and a step size of 0.01 °θ. A fixed 0.3° divergence slit as well as primary and secondary Soller slits were used. Analysis of the crystalline synthesis products was based on the LeBail method implemented in the program TOPAS Version 4.2 (Bruker-AXS 2009). Peak profiles were calculated using the fundamental parameter approach. For simulation of the background Chebychev polynomials were selected. The contribution of the amorphous phase was modeled as a single broad peak centered at 30.25 °2θ the position of which has been refined.

LeBail plot of the X-ray powder diffraction pattern of the sample retrieved from the single-crystal growth experiment. Observed step intensities are represented by grey dots. Calculated step intensities (in red) for K1.08Na0.92Ca6Si4O15 have been modelled using parameters given in Table 1 as starting values. Tick marks for the Bragg peaks of are given. The lower blue line represents the difference curve between observed and calculated step intensities. The presence of a glassy phase - indicated by an “amorphous hump” centered at about 30 °2θ - is clearly visible. (Rwp: 5.70%, Rexp: 4.77%, GOF: 1.20)
Several larger fragments of the sample were embedded in epoxy resin and polished with diamond paste (6, 3, 1 and 0.25 µm grain size) for chemical analysis and back-scattered electron (BSE) imaging using a JEOL JXA 8100 Superprobe electron probe micro-analyser (EPMA) operated in wavelength-dispersive mode with a focal-spot diameter of 2 µm. Measurements were performed with an acceleration voltage of 15 kV, a beam current of 10 nA and counting times of 20 s (peaks) and 10 s (background on each side of the peaks), respectively. For calibration, reference materials of jadeite (Na-Kα), diopside (Ca-Kα), orthoclase (K-Kα) and quartz (Si-Kα) were used. The intensities obtained were corrected for electron scattering, absorption and fluorescence radiation (ZAF correction). The BSE image of a characteristic region of the sample (Fig. 2) corroborates the observations under the optical microscope. Two phases can be clearly discriminated: light-grey partially isomorphic crystals alternate with darker grey regions representing the glassy constituents of the sample. A total number of 20 spots for each phase in various areas were analyzed. For the crystalline phase, they all yielded very similar chemical compositions giving no indication for chemical zoning with total sums based on oxides between 99.5 and 100.6 wt%. After normalization on 15 O atoms almost integral atomic ratios were obtained: K : Na : Ca : Si : O = 1.04(1) : 0.97(2) : 6.06(3) : 3.97(1) : 15 pointing to an idealized composition of KNaCa6Si4O15. The analysis of the glassy phase was more problematic as it was not very stable under the electron beam: small “burned holes” appeared on the sample surface. Therefore, total sums for the oxides varied between 96 and 98 wt%. Corresponding average values (in weight percentages) are as follows: Na2O: 10.47(6), K2O: 22.38(25), CaO: 16.15(28) and SiO2: 48.10(30).

BSE image of the sample obtained from the crystal-growth experiment. Light-grey, sometimes idiomorphically developed crystals of K1.08Na0.92Ca6Si4O15 (I) are embedded in a glassy matrix (II). Black circular regions correspond to holes produced during sample preparation
For structural investigations, a single-crystalline, transparent and colorless fragment (0.04 × 0.06 × 0.08 mm in size) of good optical quality was selected. The crystal was mounted on the tip of a glass fiber using finger-nail hardener. Data were collected on an Oxford Diffraction Gemini R Ultra single-crystal diffractometer. Intensities corresponding to one hemisphere of reciprocal space were acquired at ambient temperature (25 °C) in 342 frames with ω-scans using 0.75° scan width per frame (exposure time per frame: 75 s). Subsequent integration and data reduction were performed with the CrysAlisPRO software package (Rigaku 2015). Furthermore, a semi-empirical absorption correction (based on spherical harmonics) was applied. The diffraction pattern showed monoclinic Laue symmetry. Systematic absences pointed to space group P -21/c-. The crystal structure was solved by direct methods (program SIR2004, Burla et al. 2005). Subsequent refinement calculations were performed with the program SHELX-97 (Sheldrick 1997) embedded in the software suite WinGX (Farrugia 1999). Neutral-atom scattering coefficients and anomalous-dispersion corrections were taken from the International Tables for Crystallography, Volume C (Wilson 1995). Details of the data collection and refinement parameters are given in Table 1. Allocation of the cation species Na, K and Ca to specific sites were based on site-occupancy refinements and bond-distance considerations. Within four standard uncertainties three cation positions are exclusively occupied by calcium ions whereas the fourth site shows an almost balanced mixed potassium-sodium population (K: 54(1)%, Na: 46(1)%). Final full-matrix least-squares optimizations including fractional atomic coordinates and anisotropic displacement factors for all atoms converged to a residual value of R1 = 3.46% for 1288 independent observed reflections and 125 parameters. The largest shift/esd. in the last cycle was < 0.001. The resulting chemical composition of K1.08Na0.92Ca6Si4O15 compares well with the average composition of the crystals obtained from the EPM analysis. The final model was validated using the MISSYM algorithm included in the program PLATON (Spek 2009). No indications for unnecessarily low space group symmetry could be detected. Refined coordinates, equivalent isotropic displacement parameters, as well as selected interatomic distances and angles for K1.08Na0.92Ca6Si4O15 are summarized in Tables 2 and 3. Anisotropic displacement parameters are listed in Table 4. Figures showing structural details were prepared using the program VESTA, Version 3 (Momma and Izumi 2011).
Results
From a structural point of view, K1.08Na0.92Ca6Si4O15 belongs to the group of mixed-anion silicates containing [SiO4]- and [Si2O7]-groups in the ratio 2:1. Si-O bond distances (see Table 3) are in the normal range for silicate compounds (Liebau 1985). Furthermore, the spread of the distances is in accordance with well-established trends among oxosilicates. The bridging oxygen of the dimer resides on an inversion centre, i.e. the point group symmetry of the [Si2O7]-unit is \(\overline {1}\) (or Ci). Consequently, the Si1-O8-Si1 bond angle is straight.
Two of the three solely calcium sites are coordinated by six O atoms. The coordination polyhedra can be described as distorted octahedra (about Ca1, Ca-O bond lengths: 2.26–2.41 Å) and distorted trigonal prisms (about Ca3, Ca-O bond lengths: 2.32–2.49 Å). A single prism is linked to two adjacent octahedra by sharing a common face or edge, respectively. The resulting trimers in turn form zig-zag chains running parallel [010]. The remaining cation sites are less regularly coordinated and occupy voids of the polyhedral network. Up to 3.0 Å eight oxygen ligands can be found for each of the two sites. Whereas the M-position shows a mixed K-Na population (< M-O > = 2.778 Å) the second position is exclusively occupied by Ca2+-cations (Ca3). Bond-valence sums for the cations have been calculated using the parameters of Brown and Altermatt (1985) (for (Na,Ca,K)-O bonds) and Brese and O’Keeffe (1991) (for Si-O bonds). The results in valence units are as follows: Ca1: 2.34, Ca2: 1.75, Ca3: 1.85, Si1: 4.11 and Si2: 3.92. Notably, the calcium positions show a pronounced deviation from the ideal value of 2 v.u., indicating a significant over- or underbonding, respectively. The estimation the K-Na occupany on the mixed site from the average valence indicated an almost equal distribution of the two cation species which is in good agreement with the outcome of the site-population refinements.
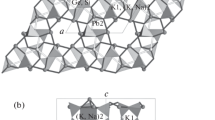
The structure can be described as a mixed tetrahedral-octahedral framework (see Fig. 3). Basic building units of this framework are so-called “kröhnkite-type” chains (named after the mineral kröhnkite, Na2Cu(SO4)2×2H2O) which are well known in inorganic chemistry (Kolitsch and Fleck 2006 and references cited therein). These chains consist of a characteristic sequence of corner-linked [SiO4]-tetrahedra and octahedra. In the present compound these chains are running parallel to [001] (Fig. 4a). The principal translation period of the kröhnkite-type chains tK of about 5.5–5.6 Å is reflected on the c lattice parameter (≈ 2 × tK) of the present compound (see Table 5). The chains are located in layers running parallel to (010) at y ≈ ¼ and ¾, respectively. In order to characterize the tetrahedral-octahedral network in more detail a topological analysis has been performed using the program TOPOS4.0 (Blatov 2012). Therefore, the crystal structure has been described by graphs composed of the vertices (sites of the tetrahedrally and octahedrally coordinated cations as well as O anions) and edges (bonds) between them. The nodes of the graph can be classified according to their coordination sequences {N k }. They represent a set of integers {N k } (k = 1,..,n), where N k is the number of sites in the kth coordination sphere of the respective atom that has been selected to be the central one. The corresponding values for the relevant symmetrically independent cation sites up to n = 10 (without the oxygen nodes) are listed in Table 6. Furthermore, the corresponding extended point symbols listing all shortest circuits for each angle for any non-equivalent atom have been determined.

Projection of the whole crystal structure of K1.08Na0.92Ca6Si4O15 parallel to [010]. [SiO4] tetrahedra and [CaO6] octahedra are shown. Small red spheres: oxygen atoms; larger blue spheres: non-octahedrally coordinated Ca-cations; bicolored spheres: mixed Na (yellow) - K (violet) M-position

Comparison of the tetrahedral-octahedral [X(SiO4)2O2]-chains in the isostructural compounds a K1.08Na0.92Ca6Si4O15 (X = Ca) and b Na2Ba6Si4O15 (X = Na). The ϕ angles at the four O-O-O hinges between the tetrahedra and octahedra have the following values (data in parentheses refer to the sodium barium silicate): ϕ1: 66.07° (90.14°), ϕ2: 103.42° (90.29°), ϕ3: 74.49° (89.38°) and ϕ4: 116.01° (90.19°)
Furthermore, for K1.08Na0.92Ca6Si4O15 the polyhedral microensembles or PME’s have been constructed. On the lowest sublevel they are formed for each octahedron and tetrahedron in the asymmetric unit by considering all directly bonded [CaO6]- and [SiO4]-groups. They represent a geometrical interpretation of the coordination sequences up to the index k = 3. The PME’s of the first sublevel observed for the Ca1 nodes can be described as follows: each [Ca1O6]-octahedron is immediately linked to six tetrahedra (Fig. 5a). Using the classification based on the calculation of the coordination sequences up to k = 3 (including the oxygen atoms) (Ilyushin and Blatov (2002)) the PME’s of Ca1 can be denoted as {6,6,18}. For the PME’s of the two crystallographically independent tetrahedral Si nodes two groups of ensembles can be distinguished: {4,3,13} (for Si1) and {4,4,20} (for Si2) (see Fig. 5b, c).

Polyhedral microensembles of the crystallographically independent octahedral (Ca1) and tetrahedral (Si) nodes: a {6,6,18} (for Ca1), b {4,3,13} (for Si1) and c {4,4,20} (for Si2)
Discussion: comparison with related structures
A comparison with other known A2B6Si4O15 compounds revealed a close similarity between the lattice parameters and space group symmetry of K1.08Na0.92Ca6Si4O15 and those reported for Na2Ba6Si4O15 (Tamazyan et al. 1987). Differences in the unit cell metrics can be attributed to differences in the sizes of divalent (Ba,Ca) and monovalent (Na,K) cations. The abovementioned resemblance pointed to an isostructural relationship which was confirmed using a graphical comparison of both crystal structures. For the calculation of several quantitative descriptors for the characterization of the degree of similarity between the crystal structures of K1.08Na0.92Ca6Si4O15 and Na2Ba6Si4O15 the program COMPSTRU (de la Flor et al. 2016) was used. Therefore, the original data of Tamazyan et al. (1987) have been transformed to monoclinic second setting. For the given two materials, the degree of lattice distortion (S), i.e. the spontaneous strain obtained from the eigenvalues of the finite Lagrangian strain tensor calculated in a Cartesian reference system, has a value of (S) = 0.0325. Subsequently, the structure of K1.08Na0.92Ca6Si4O15 was mapped on the most similar configuration of Na2Ba6Si4O15. The calculations revealed the following atomic displacements (in Å) between the corresponding atoms in the Ba- (first entry) and the Ca-(second entry) compound: Ba1-Ca3: 0.462; Ba2-Ca2: 0.087; Ba3-M1: 0.136; Na1-Ca1: 0.097; Si1-Si1: 0.175; Si2-Si2: 0.169; O1-O7: 0.301; O2-O8: 0.000; O3-O3: 0.609; O4-O2: 0.562; O5-O4: 0.310; O6-O5: 0.082; O7-O1: 0.307; O8-O6: 0.349, i.e. the maximum displacement dmax is 0.609 Å. The arithmetic mean is dav = 0.270 Å. The measure of similarity Δ as defined by Bergerhoff et al. (1999) has a value of 0.071.
The shifts between equivalent atoms in the two isostructural compounds are reflected, for example, in the conformation of the individual kröhnkite-type chains (Fig. 4a, b). As can be clearly seen, the φ angles at the four O-O-O hinges between the tetrahedra and octahedra are quite different. In Na2Ba6Si4O15 values close to 90° are observed. On the other hand, for K1.08Na0.92Ca6Si4O15 two acute (66.07, 74.49°) and two obtuse (103.42, 116.01°) angles can be distinguished. Furthermore, the [CaO6]-group in the latter compound is much more distorted than the corresponding [NaO6]-octahedron in Na2Ba6Si4O15. Unlike the Ba-containing phase, where the occurrence of twinning by reticular merohedry (mimicking an orthorhombic C-centred Bravais lattice) has been described to be a characteristic feature of almost all crystals, we could not observe similar twinning phenomena in our samples of K1.08Na0.92Ba6Si4O15.
In a recent paper on the polymorphism of Na2Ca6Si4O15, different topologies among A2B6Si4O15 phases have been compared (Kahlenberg and Maier 2016). For the ambient temperature forms K2Ca6Si4O15 and Na2Ca6Si4O15 (α-modification), two different topologies of the mixed frameworks occur (though both are based on kröhnkite-type chains). The high-temperature polymorph of Na2Ca6Si4O15 (β-phase) has the same topology as K1.08Na0.92Ca6Si4O15 and similar lattice parameters (Table 5). Furthermore, the symmetry of β-Na2Ca6Si4O15 (space group P1c1) is lower than in K1.08Na0.92Ca6Si4O15 (P121/c1). Both structures are in a group-subgroup relationship. The symmetry loss is combined with a reduction in point group symmetry and results in a translationengleiche subgroup of index 2. Based on the crystal structures of the high- and low-symmetry compounds (and neglecting differences in site populations), a symmetry-mode analysis of the global structural-distortion patterns relating β-Na2Ca6Si4O15 and K1.08Na0.92Ca6Si4O15 has been performed using the program AMPLIMODES (Perez-Mato et al. 2010). In the present case, the distortion field can be split into two contributions transforming according to the irreducible representations Γ+ 1 and Γ− 2 of P21/c. The absolute amplitudes of these two symmetry components of the global distortion are 3.03 Å (for Γ+ 1) and 4.23 Å (for Γ− 2). As can be seen from the comparison of the values, both modes have large amplitudes and both have to be considered for an understanding of the formal symmetry break, i.e. a classification into a primary and a secondary distortion is impossible.
Our results show that there is a definite influence of the monovalent cation species on the stability of different A2B6Si4O15 structure types. However, further experimental data covering a larger range of Na/K-ratios are needed to draw a final conclusion. Additionally, synthesis temperature clearly plays a role as well and, hence, in-situ diffraction studies are also of importance to detect T-induced changes in the network topologies due to phase transformations.
Finally, we would like to mention that the investigation of crystalline compounds of the system Na2O-K2O-CaO-SiO2 may have also implications for the field of Earth sciences. So far, no anhydrous sodium-potassium-calcium silicates have been observed in Nature. However, a number of water-containing minerals such as canasite, (Na,K)6Ca5Si12O30(OH,F)4, (Rastsvetaeva et al. 2003), mountainite, KNa2Ca2[Si8O19(OH)]·6H2O (Zubkova et al. 2009) or calcinaksite, KNaCa(Si4O10)·H2O (Chukanov et al. 2015) are known from various mineralogically famous regions of the Earth: the Eifel volcanic area (Germany), kimberlite pipes in Kimberley (South Africa) or different Russian localities such as the Khibiny and Lovozero plutons (Kola Peninsula) or the Murun massif (Yakutia). Therefore, it is possible that water-free variants can be found in natural environments as well. Since crystal-structure determination of the above mentioned minerals was frequently complicated due to low diffraction quality and/or complex intergrowths of different species, diffraction studies on well-crystallized synthetic counterparts may ease the identification and structural characterization of potential new minerals.
References
Arroyabe E, Kahlenberg V (2011) Structural investigations on the fertilizer component K2Ca2Si2O7. Eur J Mineral 23:101–110
Arroyabe E, Kaindl R, Többens DM, Kahlenberg V (2009a) K2Ca6Si4O15 - structural and spectroscopical studies on a mixed tetrahedral-octahedral framework. J Solid State Chem 182:3254–3261
Arroyabe E, Kaindl R, Kahlenberg V (2009b) Structural and Raman spectroscopic investigations of K4BaSi3O9 and K4CaSi3O9. Z Anorg Allg Chem 635:337–345
Bergerhoff G, Berndt M, Brandenburg K, Degen T (1999) Concerning inorganic crystal structure types. Acta Crystallogr B55:147–156
Blatov VA (2012) Nanocluster analysis of intermetallic structures with the program package TOPOS. Struct Chem 23:955–963
Brese NE, O’Keeffe MO (1991) Bond-valence parameters for solids. Acta Crystallogr B47:192–197
Brown ID, Altermatt D (1985) Bond-valence parameters obtained from a systematic analysis of the inorganic crystal structure database. Acta Crystallogr B41:244–247
Bruker-AXS (2009) TOPAS version 4.2 - software for powder diffraction data, Karlsruhe
Burla MC, Caliandro R, Camalli M, Carrozzini B, Cascarano GL, De Caro L, Giacovazzo C, Polidori G, Spagna R (2005) SIR2004: an improved tool for crystal structure determination and refinement. J Appl Crystallogr 38:381–388
Butt H, Knowles KM, Montelongo Y, Amaratunga GAJ, Wilkinson TD (2014) Devitrite-based optical diffusers. ACS Nano 8:2929–2935
Chukanov NV, Aksenov SM, Rastsvetaeva RK, Blass G, Varlamov DA, Pekov IV, Belakovskiy DI, Gurzhiy VV (2015) Calcinaksite, KNaCa(Si4O10)H2O, a new mineral from the Eifel volcanic area, Germany. Miner Petrol 109:397–404
de la Flor G, Orobengoa D, Tasci E, Perez-Mato JM, Aroyo MI (2016) Comparison of structures applying the tools available at the Bilbao Crystallographic Server. J Appl Crystallogr 49:653–664
Dilmore MF, Clark DE, Hench LL (1978) Chemical durability of Na2O-K2O-CaO-SiO2 glasses. J Am Ceram Soc 61:439–443
Farrugia LS (1999) WinGX suite for small-molecule single-crystal crystallography. J Appl Crystallogr 32:837–838
Fischer R, Tillmanns E (1984) Die Kristallstruktur von Na4CaSi3O9 und dessen strukturelle Beziehungen zu K4SrGe3O9 und Ca3Al2O6. Z Kristallogr 166:245–256 (German)
Fujikura K, Karpukhina N, Kasuga T, Brauer DS, Hill RG, Law RV (2012) Influence of strontium substitution on structure and crystallization of bioglass® 45S5. J Mater Chem 22:7395–7402
Gan FX, Cheng HS, Li QH (2006) Origin of Chinese ancient glasses—study on the earliest Chinese ancient glasses. Sci China Technol Sci 49:701–713
Grund L, Jonson B (2009) Compositional effect on fining and oxygen activity in mixed alkali silicate glass. Eur J Glass Technol B50:62–64
Ilyushin GD, Blatov VA (2002) Crystal chemistry of zirconosilicates and their analogs: topological classification of MT frameworks and suprapolyhedral invariants. Acta Crystallogr B58:198–218
Kahlenberg V, Hösch A (2002) The crystal structure of Na2Ca2Si2O7—a mixed anion silicate with defect perovskite characteristics. Z Kristallogr 217:155–163
Kahlenberg V, Maier M (2016) On the existence of a high-temperature polymorph of Na2Ca6Si4O15—implications for the phase equilibria in the system Na2O-CaO-SiO2. Miner Petrol 110:905–915
Kaygili O (2014) Synthesis and characterization of Na2O-CaO-SiO2 glass-ceramic. J Therm Anal Calorim 117:223–227
Kolitsch U, Fleck M (2006) Third update on compounds with kröhnkite-type chains: the crystal structure of wendwilsonite [Ca2Mg(AsO4)∙22H2O] and the new triclinic structure types of synthetic AgSc(CrO4)∙22H2O and M2Cu(Cr2O7)∙22H2O (M = Rb, Cs). Eur J Mineral 18:471–482
Liebau F (1985) Structural chemistry of silicates. Springer Verlag, Berlin
Lindberg D, Backman R, Chartrand P, Hupa M (2013) Towards a comprehensive thermodynamic database for ash-forming elements in biomass and waste combustion—current situation and future developments. Fuel Process Technol 105:129–141
Liu Q, Liu Y, Ding Y, Peng Z, Yu Q, Tian X, Dong G (2014) Synthesis and characterization of novel blue emitting Na2CaSiO4:Ce3+ phosphor. J Sol-Gel Sci Techn 71:276–282
Momma K, Izumi F (2011) VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J Appl Crystallogr 44:1272–1276
Perez-Mato JM, Orobengoa D, Aroyo MI (2010) Mode crystallography of distorted structures. Acta Crystallogr A66:558–590
Rastsvetaeva RK, Rozenberg KA, Khomyakov AP, Rozhdestvenskaya IV (2003) Crystal structure of F-canasite. Dokl Chem 391:177–180
Rigaku Oxford Diffraction, CrysAlisPRO software system, version 1.171.38.43 (2015) Rigaku Corporation
Schmidmair D, Kahlenberg V, Perfler L, Tribus M, Hildebrandt J, Többens DM (2015) On the ambient pressure polymorph of K2Ca3Si3O10—an unusual mixed-anion silicate and its structural and spectroscopic characterization. J Solid State Chem 228:90–98
Shelby JE (2009) Introduction to glass science and technology, 2nd edn. The Royal Society of Chemistry, Cambridge
Sheldrick GM (1997) SHELX-97—a program for crystal structure refinement. University of Göttingen, Germany
Spek AL (2009) Structure validation in chemical crystallography. Acta Crystallogr D 65:148–155
Tamazyan RA, Malinovskii A, Sirota MJ (1987) Crystal structure and twinning of Na2Ba6(Si2O7)(SiO4)2. Sov Phys Crystallogr 32:519–522
Varshneya AK (1994) Fundamentals of inorganic glasses. Academic Press Inc., San Diego
Wilson AJC (ed) (1995) International tables for crystallography. Volume C, Kluwer Academic Publishers, Dordrecht
Zubkova NV, Pekov IV, Pushcharovsky DY, Chukanov NV (2009) The crystal structure and refined formula of mountainite, KNa2Ca2[Si8O19(OH)]·6H2O. Z Kristallogr 224:389–396
Acknowledgements
Open access funding provided by University of Innsbruck and Medical University of Innsbruck. The manuscript benefitted from the helpful comments of Elena Sokolova.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editorial handling: D. Paktunc
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kahlenberg, V., Mayerl, M.JP., Schmidmair, D. et al. First investigations on the quaternary system Na2O-K2O-CaO-SiO2: synthesis and crystal structure of the mixed alkali calcium silicate K1.08Na0.92Ca6Si4O15 . Miner Petrol 112, 219–228 (2018). https://doi.org/10.1007/s00710-017-0532-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00710-017-0532-0